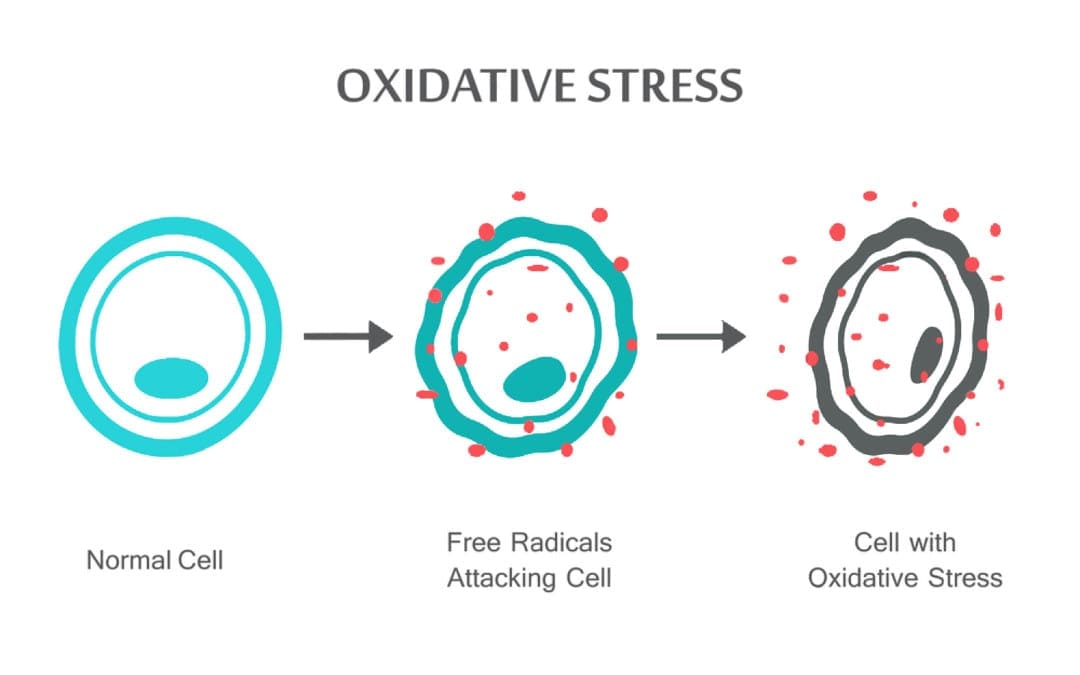
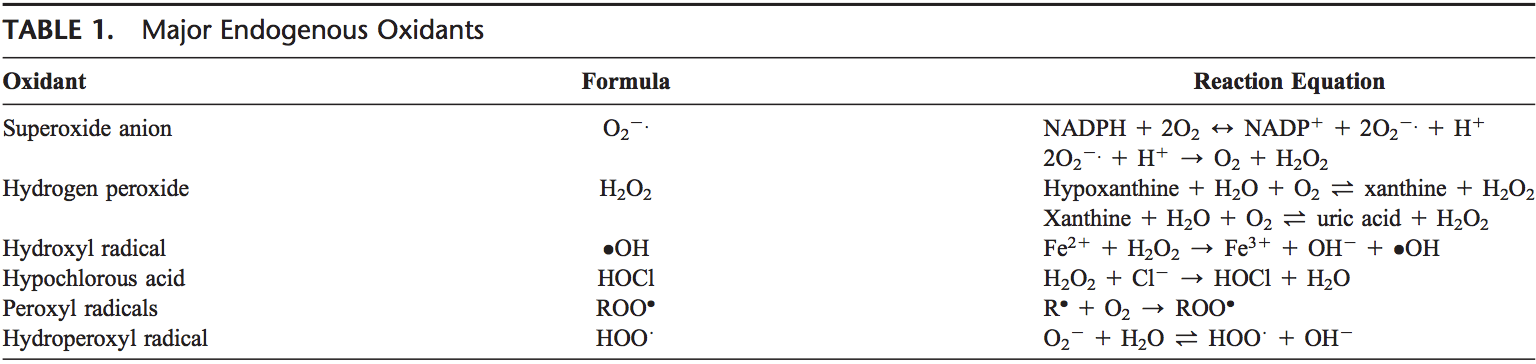
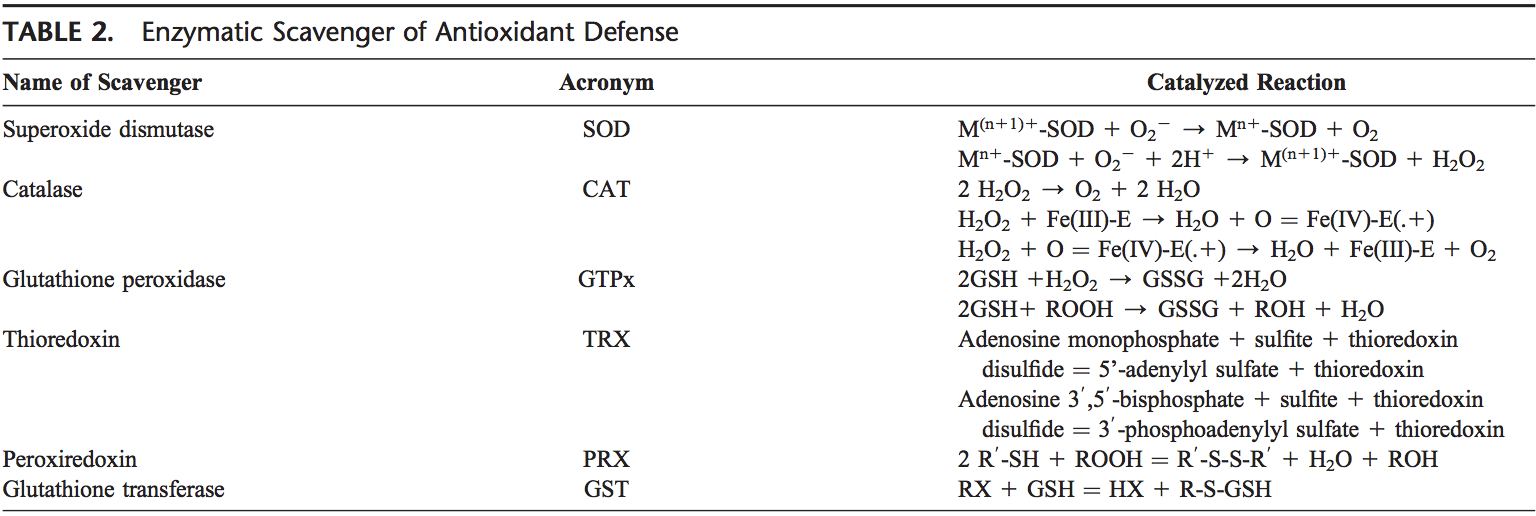
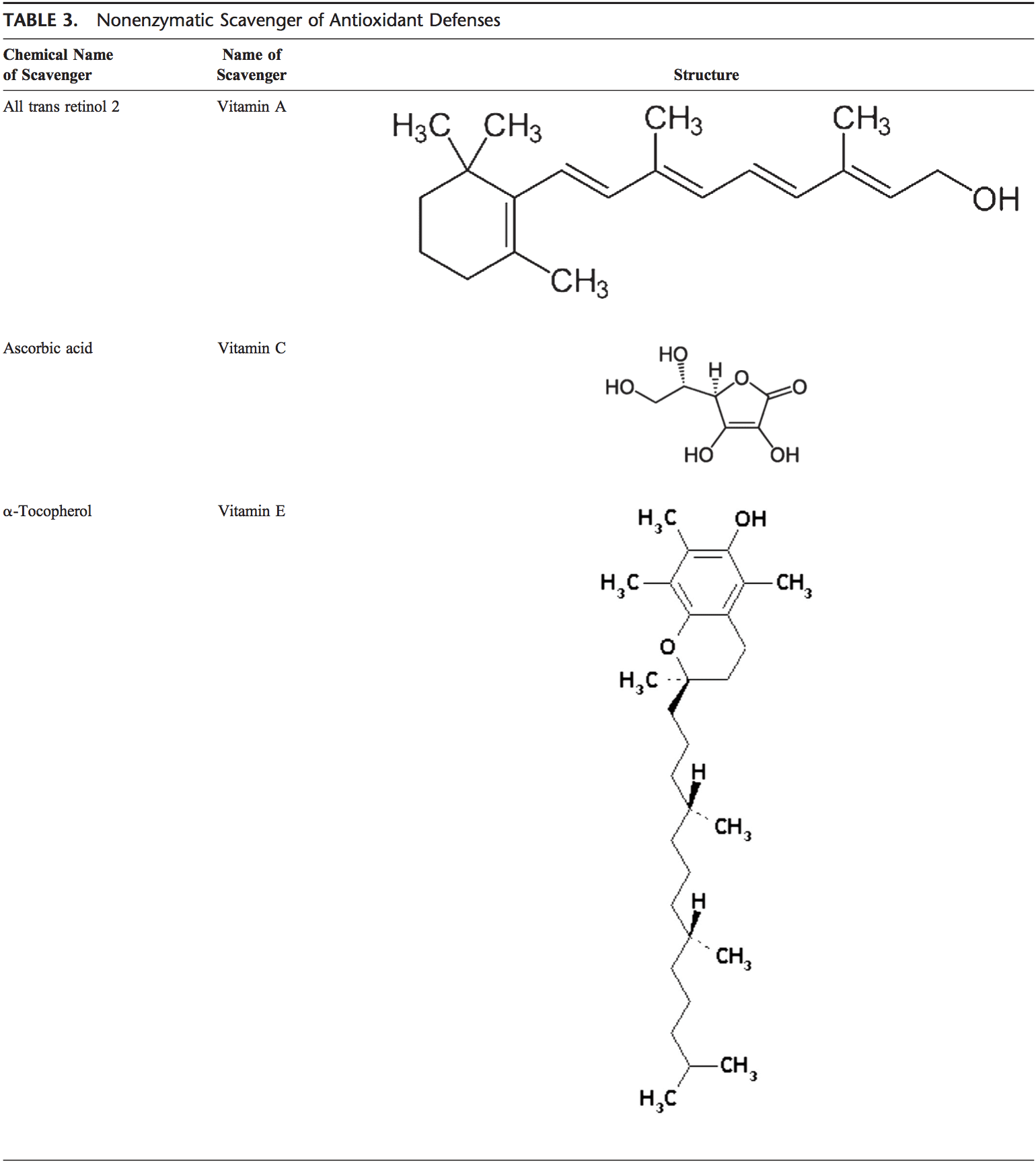
تیم پزشکی عملکردی و کایروپراکتیک استرس اکسیداتیو کلینیک برگشت. استرس اکسیداتیو به عنوان اختلال در تعادل بین تولید اکسیژن فعال (رادیکال های آزاد) و دفاع آنتی اکسیدانی تعریف می شود. به عبارت دیگر، عدم تعادل بین تولید رادیکال های آزاد و توانایی بدن برای مقابله یا سم زدایی با اثرات مضر از طریق خنثی سازی توسط آنتی اکسیدان ها است. استرس اکسیداتیو منجر به بسیاری از شرایط پاتوفیزیولوژیکی در بدن می شود. اینها عبارتند از بیماری های تخریب کننده عصبی، به عنوان مثال، بیماری پارکینسون، بیماری آلزایمر، جهش های ژنی، سرطان ها، سندرم خستگی مزمن، سندرم X شکننده، اختلالات قلب و عروق خونی، تصلب شرایین، نارسایی قلبی، حمله قلبی، و بیماری های التهابی. اکسیداسیون تحت شرایط مختلفی اتفاق می افتد:
سلول ها برای تولید انرژی از گلوکز استفاده می کنند
سیستم ایمنی مبارزه با باکتری ها و ایجاد التهاب است
بدن دفع آلودگی ها، آفت کش ها و سیگار را دود می کند
میلیون ها فرآیند در بدن ما در هر زمان خاصی رخ می دهد که می تواند باعث اکسیداسیون شود. در اینجا چند نشانه وجود دارد:
خستگی
از دست دادن حافظه و یا مه
درد عضلانی و یا مفصلی
چین و چروک همراه موی خاکستری
کاهش بینایی
سردرد و حساسیت به سر و صدا
حساسیت به عفونت
انتخاب غذاهای ارگانیک و دوری از سموم در محیط شما تفاوت بزرگی ایجاد می کند. این امر در کنار کاهش استرس، می تواند در کاهش اکسیداسیون مفید باشد.
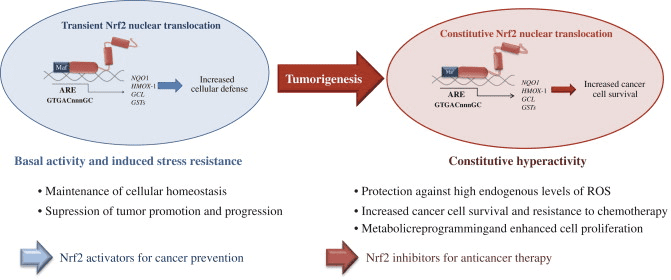
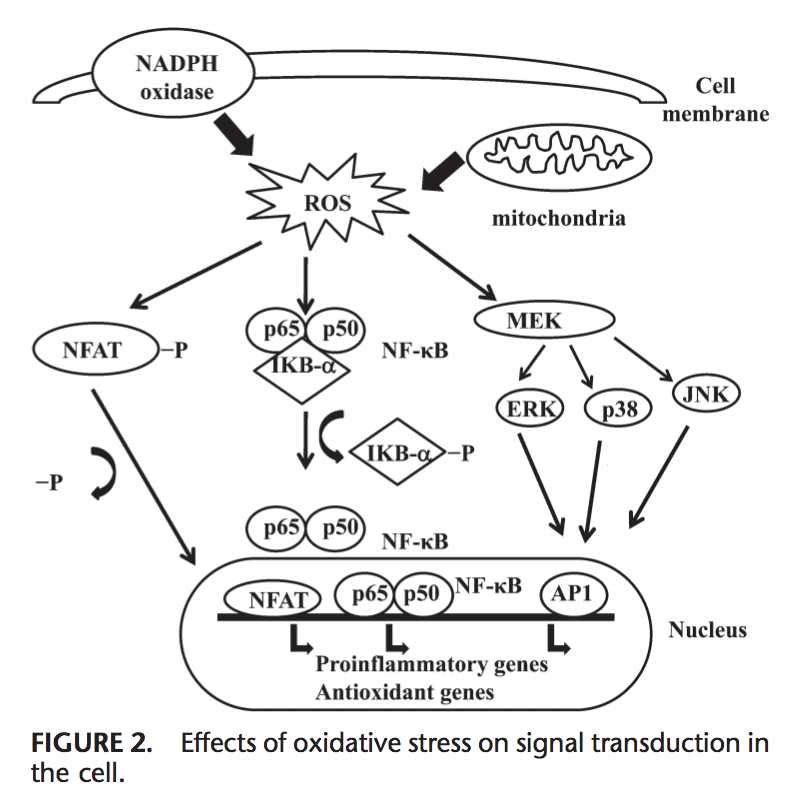
اکسیدان ها به طور کلی به منظور کنترل فرایندهای ضروری در بدن انسان، از جمله تقسیم سلولی، التهاب، عملکرد ایمنی، اتوفایگی و پاسخ استرس به طور کنترل تولید می شوند. با این حال، تولید غیرمجاز این اکسیدان ها می تواند کمک کند استرس اکسیداتیو، که ممکن است بر عملکرد سلولی تأثیر بگذارد، منجر به توسعه سمیت، بیماری مزمن و سرطان می شود. مکانیسم های آنتی اکسیدانی محافظ بدن انسان توسط یک سری مسیرهای حیاتی کنترل می شود که پاسخ سلول به اکسیدان ها را کنترل می کند. فاکتور هسته ای عامل erythroid 2، در غیر این صورت به عنوان Nrf2 شناخته می شود، یک تنظیم کننده ظهور مقاومت سلولی به اکسیدان است. هدف مقاله زیر این است که بحث و نشان دادن نقش ظهور "Nrf2" در عملکرد میتوکندری را نشان دهد.
چکیده
فاکتور رونویسی NF-E2 فاکتور 45 مربوط به p2 (Nrf2؛ نام ژن NFE2L2) با تنظیم بیان ژن شبکههای متنوع پروتئینهای محافظ سلولی، از جمله آنزیمهای آنتیاکسیدانی، ضد التهابی و سمزدایی، سازگاری و بقا در شرایط استرس را امکانپذیر میسازد. به عنوان پروتئین هایی که به ترمیم یا حذف ماکرومولکول های آسیب دیده کمک می کنند. Nrf2 با تنظیم بیوسنتز، استفاده و بازسازی گلوتاتیون، تیوردوکسین و NADPH و با کنترل تولید گونههای اکسیژن فعال توسط میتوکندری و NADPH اکسیداز، نقش مهمی در حفظ هموستاز ردوکس سلولی دارد. در شرایط هموستاتیک، Nrf2 بر پتانسیل غشای میتوکندری، اکسیداسیون اسیدهای چرب، در دسترس بودن سوبستراها (NADH و FADH2/سوکسینات) برای تنفس و سنتز ATP تأثیر می گذارد. تحت شرایط استرس یا تحریک فاکتور رشد، فعالسازی Nrf2 با افزایش تولید گونههای اکسیژن فعال در میتوکندری از طریق رونویسی رونویسی پروتئین جداکننده 3 مقابله میکند و با حفظ سطوح فاکتور 1 تنفسی هستهای و گیرنده فعالشده توسط تکثیرکننده پراکسی زوم، بیوژنز میتوکندری را تحت تأثیر قرار میدهد. coactivator 1?، و همچنین با ترویج بیوسنتز نوکلئوتید پورین. فعالکنندههای دارویی Nrf2، مانند سولفورافان ایزوتیوسیانات طبیعی، باز شدن منفذ انتقال نفوذپذیری میتوکندری و تورم میتوکندری را با واسطه اکسیدان مهار میکنند. جالب اینجاست که یک ترکیب 1,4،1,2,3-دی فنیل-2،2،XNUMX-تریازول مصنوعی، که در اصل به عنوان یک فعال کننده NrfXNUMX طراحی شده بود، برای ترویج میتوفاژی یافت شد، در نتیجه به هموستاز کلی میتوکندری کمک می کند. بنابراین، NrfXNUMX یک بازیکن برجسته در حمایت از یکپارچگی ساختاری و عملکردی میتوکندری است و این نقش به ویژه در شرایط استرس بسیار مهم است.
Nrf2 بر پتانسیل غشای میتوکندری و سنتز ATP تاثیر می گذارد.
Nrf2 بر اکسیداسیون اسید های چربی میتوكندری تاثیر می گذارد.
Nrf2 از یکپارچگی ساختاری و عملکردی میتوکندری حمایت می کند.
Activators Nrf2 اثرات مفیدی دارند وقتی که عملکرد میتوکندری به خطر افتاده است.
معرفی
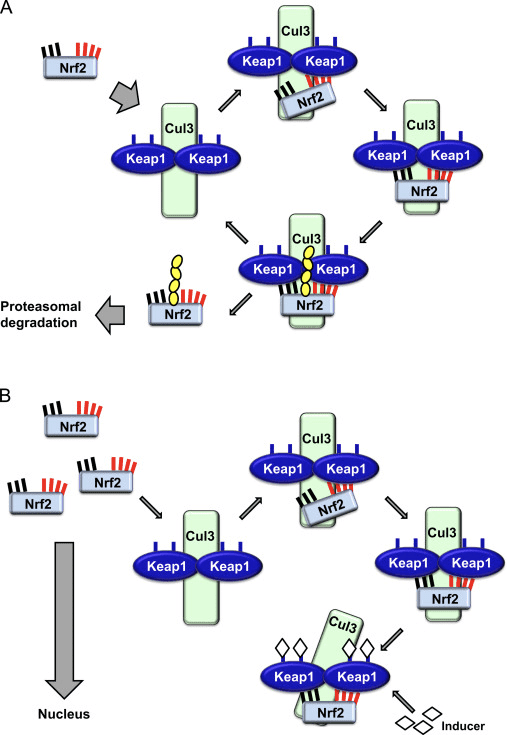
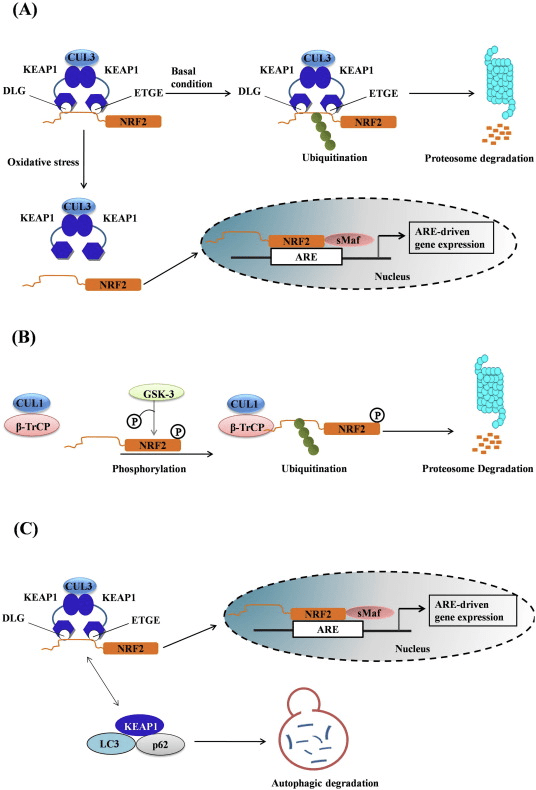
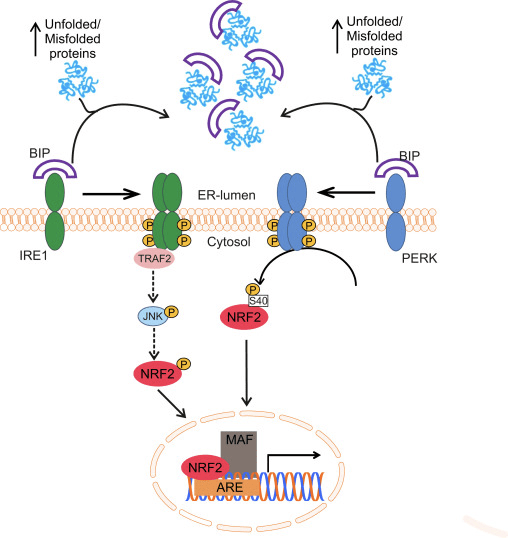
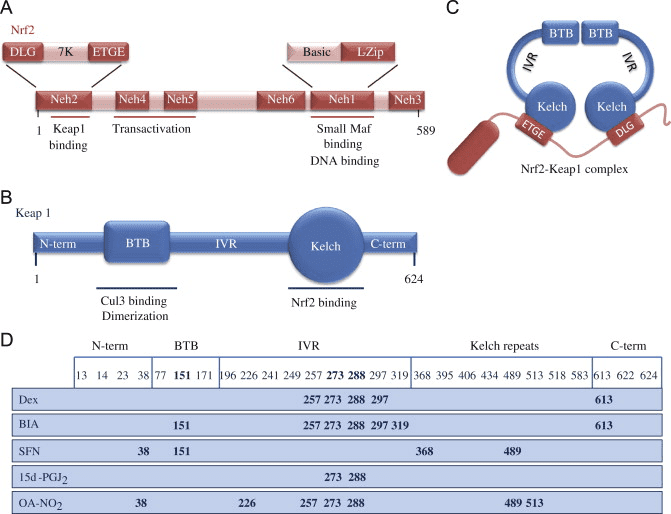
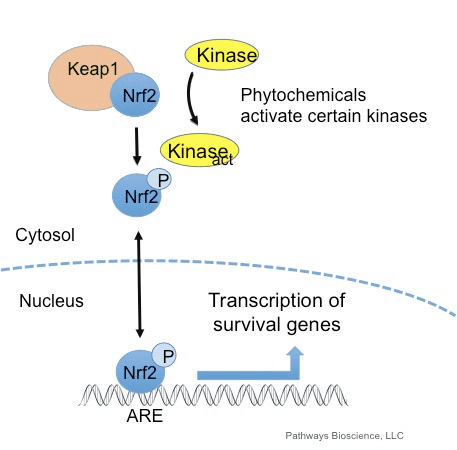
فاکتور رونویسی عامل 2 مربوط به p45 مربوط به NF-E2 (Nrf2 ؛ نام ژن NFE2L2) بیان شبکه های ژن های رمزگذار پروتئین را با فعالیت های مختلف محافظت از سلول تنظیم می کند. Nrf2 خود در درجه اول در سطح پایداری پروتئین کنترل می شود. در شرایط پایه ، Nrf2 یک پروتئین کوتاه مدت است که تحت Ubiquitination مداوم و تخریب پروتئازومی قرار می گیرد. سه سیستم شناخته شده یوبی کویتین لیگاز وجود دارد که به تخریب Nrf2 کمک می کند. از لحاظ تاریخی ، اولین تنظیم کننده منفی Nrf2 که کشف شد پروتئین 1 (Keap1) مرتبط با ECH مرتبط با کلچ بود ، یک پروتئین آداپتور سوبسترا برای کولین 1 (Cul3) / Rbx3 یوبی کویتین لیگاز [1] ، [2] ، [ 3] Keap4 از یک مکانیزم حلقوی بسیار کارآمد برای هدف قرار دادن Nrf1 برای استفاده مجدد و تخریب پروتئازومی استفاده می کند ، که در طی آن Keap2 به طور مداوم بازسازی می شود و اجازه می دهد چرخه ادامه یابد (شکل 1A) [1]. Nrf5 نیز با واسطه گلیکوژن سنتاز کیناز (GSK) 2 /؟ - وابسته به TrCP لیباز یوبی کویتین مبتنی بر Cul3 در معرض تخریب قرار می گیرد [1] ، [6]. اخیراً ، گزارش شده است که ، در شرایط تنش شبکه آندوپلاسمی ، Nrf7 در یک فرآیند واسطه توسط E2 یوبی کویتین لیگاز Hrd3 ، مجدداً تجزیه و تخریب می شود [1].
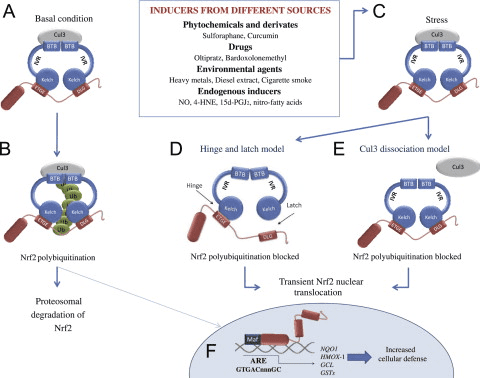
شکل 1 مدل پیوستگی و بازسازی چرخه ای برای تخریب ناشی از Keap1 از Nrf2. (A) Nrf2 به طور پیوسته به یک dimer رایگان Keap1 متصل می شود: ابتدا از طریق اتصال دامنه اتصال ETGE (سرخ) با سرعتی بالا و سپس از طریق دامنه اتصال DLG (چهره های سیاه) با کمترین وابستگی. در این ترکیب پروتئین پیچیده، Nrf2 تحت ubiquitination تحت آزمایش قرار می گیرد و برای تخریب پروتئازوما هدف قرار می گیرد. رایگان Keap1 بازسازی شده است و قادر به اتصال به Nrf2 تازه ترجمه شده است، و این چرخه دوباره شروع می شود. B) Inducers (الماس سفید) با سیگنال های سنسور Keap1 واکنش نشان می دهند (منجر به تغییرات سازنده ای شده و باعث کاهش آسیب پذیری بستر می شود). Keap1 رایگان بازسازی نشده است، و Nrf2 به تازگی synthesized تجمع و انتقال به هسته.
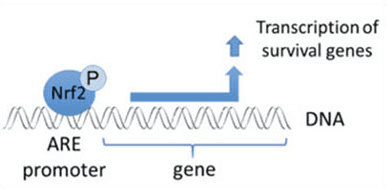
Keap1 علاوه بر خدمت به عنوان یک پروتئین آداپتور زیربخش لیگاز ubiquitin ligase، حسگر برای مجموعه ای گسترده از فعال کننده های کوچک مولکولی Nrf2 (نامگذاری کننده نامیده می شود) [9] است. Inducers بلوک چرخه تخریب ناشی از Keap1 از Nrf2 را با تغییر شیمیایی بقایای خاص سیتستین در Keap1 [10]، [11] و یا به طور مستقیم از بین بردن رابط اتصال Keap1: Nrf2 [12]، [13] را مسدود می کند. در نتیجه، Nrf2 تضعیف نمی شود و فاکتور رونویسی تجمع می یابد و به هسته منتقل می شود (شکل 1B)، جایی که آن را یک heterodimer با یک پروتئین Maf کوچک تشکیل می دهد؛ به عناصر واکنش آنتی اکسیدانی، مناطق تنظیم کننده بالادست از ژن های هدف آن متصل می شود؛ و آغاز رونویسی [14]، [15]، [16]. باتری از اهداف Nrf2 شامل پروتئین هایی با عملکرد متفاوت سیتوپروتئینی، از جمله آنزیم های متابولیسم زنجبیل، پروتئین هایی با عملکرد آنتی اکسیدان و ضد التهابی و زیربخش های پروتئازوما و پروتئین هایی است که هومیوستیس سرمی سلول را تنظیم می کنند و در متابولیسم واسطه ای شرکت می کنند.
Nrf2: یک رگولاتور استاد ردوکسوموستازیک سلولی
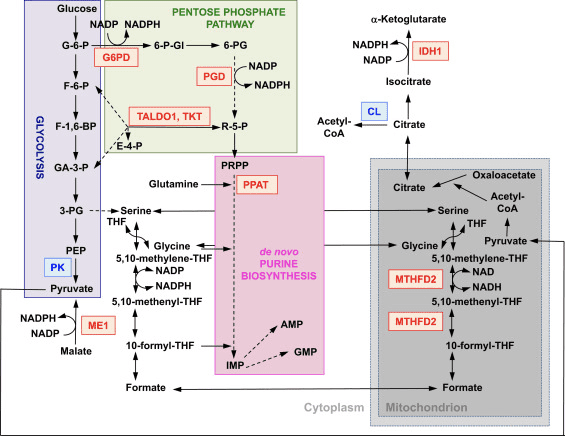
عملکرد Nrf2 به عنوان یک تنظیم کننده اصلی هموستاز ردوکس سلولی به طور گسترده ای شناخته شده است. بیان ژن هر دو زیرواحد کاتالیزوری و تنظیمی ب-گلوتامیل سیستئین لیگاز، آنزیمی که مرحله محدود کننده سرعت در بیوسنتز گلوتاتیون کاهش یافته (GSH) را کاتالیز می کند، مستقیماً توسط Nrf2 تنظیم می شود [17]. زیرواحد xCT سیستم xc-، که سیستین را به سلولها وارد میکند، همچنین هدف رونویسی مستقیم Nrf2 است [18]. در سلول، سیستین به سیستئین تبدیل می شود که پیش ماده ای برای بیوسنتز GSH است. علاوه بر نقش خود در بیوسنتز GSH، Nrf2 وسیله ای را برای حفظ گلوتاتیون در حالت کاهش یافته با تنظیم رونویسی هماهنگ گلوتاتیون ردوکتاز 1 [19]، [20] فراهم می کند که با استفاده از معادل های احیا کننده از NADPH، گلوتاتیون اکسید شده را به GSH کاهش می دهد. . NADPH مورد نیاز توسط چهار آنزیم اصلی مولد NADPH، آنزیم مالیک 1 (ME1)، ایزوسیترات دهیدروژناز 1 (IDH1)، گلوکز-6-فسفات دهیدروژناز (G6PD) و 6-فسفوگلوکونات دهیدروژناز (PGD) تامین می شود. به طور رونویسی تا حدی توسط Nrf2 (شکل 2) تنظیم شده است [21]، [22]، [23]، [24]. جالب اینجاست که Nrf2 همچنین بیان ژن القایی اشکال سیتوزولی، میکروزومی و میتوکندری آلدهید دهیدروژناز [25] را تنظیم می کند که از NAD(P)+ به عنوان کوفاکتور استفاده می کند و باعث ایجاد NAD(P)H می شود. در واقع، سطوح NADPH و نسبت NADPH/NADP+ در فیبروبلاستهای جنینی جدا شده از موشهای Nrf2-knockout (Nrf2-KO) در مقایسه با سلولهای همتایان نوع وحشی (WT) آنها کمتر است و سطوح NADPH با ناکداون Nrf2 کاهش مییابد. رده های سلولی سرطانی با Nrf2 فعال اصلی [26]. همانطور که انتظار می رود، سطوح GSH در سلول هایی که Nrf2 در آنها مختل شده است کمتر است. برعکس، فعالسازی Nrf2 با روشهای ژنتیکی یا دارویی منجر به افزایش تنظیم GSH میشود [27]، [28]، [29]. نکته مهم این است که Nrf2 بیان ژن تیوردوکسین [30]، [31]، [32]، تیوردوکسین ردوکتاز 1 [28]، [29]، [32]، [33] و سولفیردوکسین [34] را که ضروری هستند، تنظیم می کند. برای کاهش تیول های پروتئین اکسید شده
شکل 2 نقش Nrf2 در متابولیسم سلولهای به سرعت در حال تکثیر. Nrf2 یک تنظیم کننده مثبت برای ژنهای رمز کننده آنزیمها در بازوی اکسیداتیو است (به عنوان مثال: گلوکز-6-فسفات دهیدروژناز (G6PD) و 6-فسفوگلوبونات دهیدروژناز (PGD)] و بازوی غیر اکسیداتیو [یعنی ترانسالدولاز 1 (TALDO1) و ترانسکتولاز ( TKT)] از مسیر پنتوز فسفات. G6PD و PGD NADPH تولید می کنند. Nrf2 همچنین بیان ژن دو آنزیم مولد NADPH دیگر ، آنزیم مالیک 1 (ME1) و ایزوسیترات دهیدروژناز 1 (IDH1) را تنظیم می کند. بیان ژنی آمیدوترانسفراز فسفریبوسیل پیرو فسفات (PPAT) ، که ورود به مسیر بیوسنتز پورین de novo را کاتالیز می کند ، همچنین به عنوان بیان متیلن تتراهیدروفولات دهیدروژناز 2 (MTHFD2) ، یک آنزیم میتوکندری با یک نقش مهم توسط Nrf2 تنظیم می شود ارائه واحدهای تک کربنی برای بیوسنتز پورین de novo. پیروات کیناز (PK) توسط Nrf2 تنظیم منفی می شود و انتظار می رود که از ایجاد واسطه های گلیکولیتیک و همراه با G6PD کانال متابولیت از طریق مسیر پنتوز فسفات و سنتز اسیدهای نوکلئیک ، اسیدهای آمینه و فسفولیپیدها پشتیبانی کند. Nrf2 بیان ژنی ATP-citrate lyase (CL) را تنظیم می کند ، که ممکن است در دسترس بودن سیترات برای استفاده از میتوکندری یا (از طریق isocitrate) برای IDH1 را افزایش دهد. قرمز و آبی به ترتیب تنظیم مثبت و منفی را نشان می دهند. میتوکندری به رنگ خاکستری نشان داده شده است. اختصارات متابولیت: G-6-P ، گلوکز 6-فسفات ؛ F-6-P ، فروکتوز 6-فسفات ؛ F-1,6،1,6-BP ، فروکتوز 3،3-بیس فسفات ؛ GA-3-P ، گلیسرآلدئید 3-فسفات ؛ 6-PG ، 6-فسفوگلیسیرات ؛ PEP ، فسفنول پیروات ؛ 6-P-Gl ، 6-phosphogluconolactone ؛ 5-PG ، 5-فسفوگلوكونات ؛ R-5-P ، 1-فسفات ریبولوز ؛ PRPP ، XNUMX-فسفریبوسیل -؟ - XNUMX-پیرو فسفات ؛ THF ، تتراهیدروفولات ؛ IMP ، مونوفسفات اینوزین ؛ AMP ، آدنوزین مونوفسفات ؛ GMP ، مونوفسفات گوانوزین.
با توجه به نقش حیاتی Nrf2 به عنوان یک تنظیم کننده اصلی تنظیم کننده هومئوستاز سرطانی سلولی، شگفت آور نیست که در مقایسه با سلول های WT، سطوح گونه های اکسیژن واکنشی (ROS) در سلول هایی که در آنها Nof2 خراب شده است (Nrf2-KO) [35] این تفاوت به ویژه در مورد چالش با عوامل ایجاد کننده استرس اکسیداتیو موثر است. علاوه بر این، سلولهای کمبودی در Nrf2 حساسیت بیشتری نسبت به سمیت اکسیدانهای مختلف دارند و توسط القایی Nrf2 که تحت شرایط مشابه محافظت از سلولهای WT [29]، [36] ، [37]. علاوه بر کلسترول سرمی سلولهای سرطانی، Nof2 نیز برای نگهداری هوموتازیست سرم درمانی میتوکندری بسیار مهم است. بنابراین، در مقایسه با WT، کل NADH میتوکندری به طور قابل توجهی افزایش یافته در Keap1-KO و به طور چشمگیری در سلولهای Nrf2-KO [35] کاهش می یابد.
با استفاده از تصویربرداری سلول زنده، ما اخیرا میزان تولید ROS را در کولوکورهای گلونیورونال اولیه و برشهای بافت مغزی جدا شده از WT، Nrf2-KO یا Keap1-knockdown (Keap1-KD) [38] جدا کردیم. همانطور که انتظار می رفت، میزان تولید ROS در سلول ها و بافت های Nrf2-KO سریع تر بود در مقایسه با همتایان WT. با این حال، مشاهدات غیرمنتظره ای که در مقایسه با WT انجام دادیم، سلول های Keap1-KD نیز دارای میزان بالایی از تولید ROS بودند، هرچند که تفاوت بین ژنوتیپ های WT و Keap1-KD کمتر از WT و Nrf2-KO بود . سپس ماژولهای mRNA NOX2 و NOX4، زیرمجموعه کاتالیزوری دو ایزوفرم NADPH اکسیداز (NOX) را که در پاتولوژی مغز رخ داده است، مورد بررسی قرار دادیم و دریافتیم که NOX2 به طور چشمگیری در شرایط کمبود Nrf2 افزایش می یابد، در حالی که NOX4 تنظیم می شود وقتی که Nrf2 اگر چه به میزان کمتری فعال است. به طور كلی، میزان بالا بردن سلولها و بافتهای موشهای جهش یافته با افزایش تولید مشابه در تولید ROS [38] همراه است. جالب توجه است، Nrf2 نه تنها NADPH اکسیداز را تنظیم می کند، بلکه ROS تولید شده توسط NADPH اکسیداز می تواند Nrf2 را فعال کند، همانطور که در سلول های اپیتلیال ریه و کراتومیوسیت ها [39]، [40] نشان داده شده است. علاوه بر این، یک مطالعه اخیر نشان داده است که فعال سازی NADPH اکسیداز وابسته به Nrf2 مکانیسم مهمی برای محافظت در برابر آسیب های میتوکندری و مرگ سلولی در قلب در هنگام اضافه بار اضافی فشار [41] می باشد.
علاوه بر فعالیت کاتالیزوری NADPH اکسیداز، تنفس میتوکندری یکی دیگر از منابع اصلی درون سلولی ROS است. با استفاده از پروتئین MitoSOX اختصاصی میتوکندری، ما نقش ROS منشاء میتوکندری را به تولید کلی ROS در کولوکولاتورهای گلونیورونال اولیه جدا از WT، Nrf2-KO، یا موشهای Keap1-KD [38]. همانطور که انتظار می رفت، سلول های Nrf2-KO دارای میزان بالاتر تولید ROS میتوکندری نسبت به WT بودند. با توافق با یافته های تولید کلی ROS، میزان تولید ROS میتوكندری در Keap1-KD نیز نسبت به سلول های WT بالاتر بود. مهم است که مسدود کردن پیچیده I با راتنون موجب افزایش قابل توجهی در تولید ROS میتوكندری در سلولهای WT و Keap1-KD شد، اما در cells Nrf2-KO اثر نداشت. در مقایسه با افزایش انتظار می رود تولید ROS میتوکندری در سلول های WT پس از اضافه کردن پیروات (برای افزایش دسترسی به NADH، افزایش پتانسیل غشای میتوکندری و طبیعی شدن تنفس) تولید ROS در سلول های Nrf2-KO کاهش می یابد. با هم، این یافته ها به شدت نشان می دهد که در غیاب Nrf2: (1) فعالیت پیچیده I ضعیف است، (2) فعالیت ناشی از پیچیده I به دلیل محدودیت های زیربناها، و (3) فعالیت ناشی از پیچیده من یکی از دلایل اصلی افزایش تولید ROS میتوكندری است كه احتمالا به علت جریان برگشتی الکترون از پیچیده II است.
Nrf2 بر روی پتانسیل غشای میتوکندری و تنفس تاثیر می گذارد
پتانسیل غشای میتوکندری (؟؟ متر) شاخص جهانی سلامت میتوکندری و وضعیت متابولیکی سلول است. در یک سلول سالم ، ؟؟ متر توسط زنجیره تنفسی میتوکندری حفظ می شود. جالب توجه است ، یک برچسب گذاری ایزوتوپی پایدار با اسیدهای آمینه در مطالعه پروتئومیکس مبتنی بر فرهنگ در سلول سلول MCF10A اپیتلیال پستان انسان گیرنده منفی استروژن نشان داده است که جز component زنجیره انتقال الکترون میتوکندری با فعال سازی دارویی (توسط سولفورافان) Nrf4 تنظیم می شود ، در حالی که تعدیل ژنتیکی Nrf2 (با استفاده از Knapdown Keap2) منجر به کاهش تنظیم زیرواحدهای اکسیداز سیتوکروم c COX1 و COX2I4 می شود [1]. مطالعه پروتئوم کبد با استفاده از الکتروفورز ژل دو بعدی و طیف سنجی جرمی دفع لیزر / یونیزاسیون با کمک ماتریس نشان داده است که Nrf42 بیان زیر واحد ATP سنتاز را تنظیم می کند؟ [2] علاوه بر این ، گزارش شده است که پروتئین میتوکندری DJ-43 ، که در حفظ فعالیت کمپلکس I [1] نقش دارد ، برای تثبیت Nrf44 [2] ، [45] گزارش شده است ، اگرچه اثرات محافظت نورونی فعال سازی دارویی یا ژنتیکی از Nrf46 مستقل از DJ-2 هستند [1]. با این حال ، عواقب این مشاهدات برای عملکرد میتوکندری بررسی نشده است.
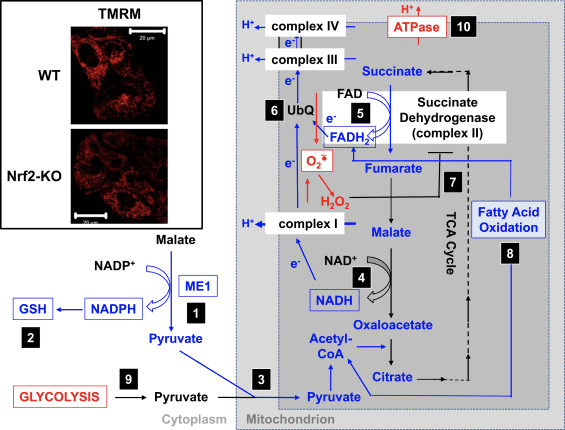
در توافق با فعالیت مختل I پیچیده در شرایط کمبود Nrf2 ، پایه پایه در Nbf2-KO فیبروبلاست جنینی موش (MEF) و سلولهای اولیه Glioneuronal کشت در مقایسه با همتایان WT کمتر است (شکل 3 ، سوراخ) [35] در مقابل ، هنگامی که Nrf2 از نظر ژنتیکی از نظر ژنتیکی تنظیم مجدد می شود (با ناک اوون یا ناک اوت Keap1) پایه متر بالاتر است. این تفاوت در متر در ژنوتیپ ها نشان می دهد که تنفس تحت تأثیر فعالیت Nrf2 قرار دارد. در واقع ، ارزیابی میزان مصرف اکسیژن در حالت پایه نشان داده است که ، در مقایسه با WT ، مصرف اکسیژن در MEF های Nrf2-KO و Keap1-KO به ترتیب 50 and و ~ 35 lower کمتر است.
شکل 3 مکانیزم پیشنهادی برای تضعیف عملکرد میتوکندری در شرایط کمبود Nrf2. (1) کاهش سطح ME1 ، IDH1 ، G6PD و PGD منجر به کاهش سطح NADPH می شود. (2) سطح GSH نیز پایین است. (3) فعالیت کم ME1 ممکن است استخر پیروات وارد شده به میتوکندری را کاهش دهد. (4) تولید NADH کندتر است و منجر به اختلال در فعالیت کمپلکس I و افزایش تولید ROS میتوکندری می شود. (5) کاهش FAD به FADH2 در پروتئین های میتوکندری نیز کاهش یافته و جریان الکترون را از FADH2 به UbQ کاهش داده و به کمپلکس III می رساند. (6) تشکیل کندتر UbQH2 ممکن است فعالیت آنزیمی سوکسینات دهیدروژناز را کاهش دهد. (7) افزایش سطح ROS ممکن است فعالیت کمپلکس II را بیشتر مهار کند. (8) بازده پایین اکسیداسیون اسیدهای چرب به کاهش در دسترس بودن بستر برای تنفس میتوکندری کمک می کند. (9) گلیکولیز به عنوان یک مکانیسم جبرانی برای کاهش تولید ATP در فسفوریلاسیون اکسیداتیو افزایش می یابد. (10) سنتاز ATP برای حفظ ؟؟ متر عمل می کند. قرمز و آبی به ترتیب تنظیم مجدد و تنظیم مقررات را نشان می دهند. این جعبه ها نشانگر در دسترس بودن شواهد تجربی است. این حصار تصاویری از میتوکندری آستروسیتهای قشر WT و Nrf2-KO را که توسط پروب فلورسنت تترامتیل رودامین متیل استر تجسم یافته اند نشان می دهد (TMRM ؛ 25 نانومتر). نوار مقیاس ، 20 دقیقه.
این تفاوت در مترمربع و تنفس در بین ژنوتیپ ها با میزان استفاده از بسترها برای تنفس میتوکندری منعکس می شود. استفاده از بسترها برای چرخه اسید تری کاربوکسیلیک (TCA) (مالات / پیروات ، که به نوبه خود تولید کمپلکس I بستر NADH را افزایش می دهد) یا متیل سوکسینات ، یک بستر برای کمپلکس II ، باعث افزایش گام به گام میلی متر در هر دو WT می شود و سلولهای عصبی Keap1-KD ، اما میزان افزایش در سلولهای Keap1-KD بیشتر است. از همه مهمتر ، اشکال پاسخ به این بسترهای چرخه TCA بین دو ژنوتیپ متفاوت است ، به موجب آن افزایش سریع ؟؟ متر در سلولهای Keap1-KD با افزودن سوبسترا به جای فلات با یک افت سریع دنبال می شود ، که نشان می دهد یک غیر معمول است مصرف سریع بستر این یافته ها با سطوح بسیار پایین (50-70٪) مالات ، پیروات و سوکسینات که پس از یک نبض 1 ساعته [U-13C6] گلوکز در Keap1-KO در مقایسه با WT MEF مشاهده شده است ، کاملاً مطابقت دارد. سلولها [24]. در نورون های Nrf2-KO ، فقط پیروات قادر به افزایش m متر است ، در حالی که مالات و متیل سوکسینات باعث دپلاریزاسیون خفیف می شوند. به نظر می رسد اثر Nrf2 بر تولید بستر میتوکندری مکانیسم اصلی است که Nrf2 بر عملکرد میتوکندری تأثیر می گذارد. شاخص ردوکس NADH میتوکندری (تعادل بین مصرف NADH توسط کمپلکس I و تولید NADPH در چرخه TCA) در سلول های Nrf2-KO در مقایسه با نمونه های WT آنها به طور قابل توجهی پایین تر است ، و علاوه بر این ، میزان بازسازی استخرهای NADH و FADH2 پس از مهار کمپلکس IV (با استفاده از NaCN) در سلولهای جهش یافته کندتر هستند.
در میتوکندری جدا شده از مغز و کبد موش ، مکمل بسترها برای کمپلکس I یا برای کمپلکس II میزان اکسیژن مصرفی را هنگام فعال شدن Nrf2 با شدت بیشتری افزایش می دهد و وقتی Nrf2 مختل می شود با بازده کمتری کار می کند. بنابراین ، مالات میزان بالاتری از مصرف اکسیژن را در Keap35-KD در مقایسه با WT ایجاد می کند ، اما اثر آن در Nrf1-KO میتوکندری ضعیف تر است. به طور مشابه ، در حضور روتنون (هنگامی که کمپلکس I مهار می شود) ، سوکسینات در مقایسه با WT در Keap2-KD میزان اکسیژن را بیشتر فعال می کند ، در حالی که پاسخ در میتوکندری های Nrf1-KO کاهش می یابد. علاوه بر این ، فرهنگهای عصبی اولیه Nrf2-KO و موشها نسبت به سمیت مهارکننده های کمپلکس II 2-نیتروپروپیونیک اسید و مالونات حساس تر هستند ، در حالی که پیوند داخل بینی آستروسیتهای ابراز کننده زیاد Nrf3 محافظ است [2] ، [48]. به همین ترتیب ، موش های Nrf49-KO حساسیت بیشتری نشان می دهند ، در حالی که فعال سازی ژنتیکی یا دارویی Nrf2 دارای اثرات محافظتی در برابر مسمومیت عصبی ناشی از یون 2-متیل -1-فنیل پیریدینیوم مهارکننده پیچیده I در 4-متیل-1-فنیل-4 است مدل حیوانی 1,2,3,6،49،50-tetrahydropyridine بیماری پارکینسون [51] ، [52] ، [53] ، [54] ، [55] ، [56] ، [57] ، [58] ، [59] ، [60] ، [61] ، [XNUMX] ، [XNUMX].
نسبت کنترل تنفسی (RCR) ، نسبت حالت 3 (تحریک شده توسط ADP) به حالت تنفس 4 (بدون وجود ADP) ، در غیاب Nrf2 کاهش می یابد ، اما RCR بین Keap1-KD و WT میتوکندری مشابه است [35 ] از آنجا که RCR نشانه ای از درجه اتصال فعالیت زنجیره تنفسی میتوکندری به فسفوریلاسیون اکسیداتیو است ، این یافته نشان می دهد که میزان تنفس بالاتر در میتوکندری Keap1-KD به دلیل جدا شدن فسفوریلاسیون اکسیداتیو نیست. این همچنین نشان می دهد که فسفوریلاسیون اکسیداتیو هنگام فعال شدن Nrf2 کارآمدتر است. میزان تنفس بالاتر در میتوکندری Keap1-KD با سطوح بالاتر تولید ROS میتوکندری سازگار است [38] زیرا سرعت تنفس بالاتر ممکن است منجر به افزایش نشت الکترون شود. با این حال ، تحت شرایط استرس اکسیداتیو ، افزایش تولید ROS توسط تنظیم رونویسی وابسته به Nrf2 پروتئین اتصال 3 (UCP3) خنثی می شود ، که باعث افزایش هدایت پروتون غشای داخلی میتوکندری و در نتیجه تولید سوپراکسید می شود [62]. به تازگی ، نشان داده شده است که محصول پراکسیداسیون لیپید 4-هیدروکسی-2-غیر کلیه باعث تعدیل وابسته به Nrf2 در UCP3 در قلب می شود. این ممکن است به ویژه برای محافظت در شرایط استرس اکسیداتیو از جمله مواردی که در طی ایسکمی مجدد جریان خون وجود دارد [63].
Nrf2 بر کارایی فسفوریلاسیون اکسیداتیو و سنتز ATP تاثیر می گذارد
در توافق با اثر Nrf2 بر تنفس ، در میتوکندری مغز و کبد ، کمبود Nrf2 منجر به کاهش کارایی فسفوریلاسیون اکسیداتیو می شود (همانطور که با نسبت ADP به اکسیژن مصرف می شود ، که برای سنتز ATP مصرف می شود) ، در حالی که فعال شدن Nrf2 (Keap1) -KD) اثر معکوس دارد [35]. در مقایسه با WT ، سطح ATP در سلولهای دارای تنظیم مجدد سازنده Nrf2 به طور قابل توجهی بالاتر است و وقتی Nrf2 سرنگون شود [64] یا مختل می شود ، کمتر است. علاوه بر این ، استفاده از مهارکننده های فسفوریلاسیون اکسیداتیو (الیگومایسین) یا گلیکولیز (اسید یدو استیک) نشان داده است که Nrf35 نحوه تولید سلولهای ATP را تغییر می دهد. بنابراین ، در سلول های عصبی WT ، الیگومایسین باعث کاهش کامل ATP می شود و اسید یدواستیک اثر دیگری ندارد. نکته قابل توجه ، در سلولهای Nrf2-KO ، الیگومایسین باعث افزایش سطح ATP می شود که سپس به تدریج ، اما به طور کامل توسط اسید یدواستیک تخلیه می شود ، این نشان می دهد که در غیاب Nrf2 ، گلیکولیز و نه فسفوریلاسیون اکسیداتیو ، منبع اصلی تولید ATP است. جالب توجه است که ، با وجود افزایش کارایی فسفوریلاسیون اکسیداتیو در سلولهای Keap2-KD ، افزودن اولیگومایسین منجر به کاهش٪ 1 in در سطح ATP می شود و اسید یدواستیک باعث کاهش ~ 80 further بیشتر می شود. بنابراین ، کمبود Nrf20 یا فعال سازی سازنده آن باعث کاهش سهم فسفوریلاسیون اکسیداتیو و افزایش سهم گلیکولیز در سنتز ATP می شود. این اثر به ویژه هنگامی که Nrf2 غایب است آشکار می شود و با وابستگی ؟؟ متر به حضور گلوکز در محیط [2] و افزایش سطح واسطه های گلیکولیتیک سازگار است (G-35-P ، F-6-P ، دی هیدروکسی استون فسفات ، پیروات و لاکتات) پس از حذف Nrf6 [2].
افزایش سطح ATP پس از مهار F1F0-ATPase توسط اولیگومایسین نشان می دهد که در غیاب Nrf2 ، F1F0-ATPase به عنوان یک ATPase و نه یک سنتاز ATP عمل می کند ، یعنی برعکس عمل می کند. چنین واژگونی در فعالیت به احتمال زیاد نشان دهنده نیاز به پمپاژ پروتونها از طریق غشای داخلی میتوکندری در تلاش برای حفظ متر است ، که برای یکپارچگی عملکردی این اندامک بسیار مهم است. برگشت عملکرد F1F0-ATPase نیز با دپلاریزاسیون میتوکندری مشاهده شده بر روی تجویز اولیگومایسین به سلولهای Nrf2-KO مشهود است ، که در تضاد شدید با افزایش قطبش در همتایان کمبود WT یا Keap1 است [35]. به طور کلی ، به نظر می رسد که در شرایط کمبود Nrf2 ATP در درجه اول در گلیکولیز تولید می شود ، و سپس این ATP تا حدی توسط F1F0-ATPase برای حفظ ؟؟ متر استفاده می شود.
Nrf2 باعث افزایش اکسیداسیون چربی های میتوکندری می شود
اثر کمبود Nrf2 بر روی ؟؟ متر به ویژه هنگامی برجسته می شود که سلولها در محیطی بدون گلوکز انکوبه شوند و ؟؟ m در Nrf50-KO٪ 2٪ کمتر از سلولهای WT است [35]. در شرایط محرومیت از گلوکز ، اکسیداسیون اسیدهای چرب میتوکندری (FAO) تأمین کننده اصلی بسترها برای تنفس و فسفوریلاسیون اکسیداتیو است ، که نشان می دهد Nrf2 ممکن است بر FAO تأثیر بگذارد. در حقیقت ، کارایی FAO برای اسید چرب اشباع شده با زنجیره بلند (C16: 0) اسید پالمیتیک و هگزانوئیک اسید با زنجیره کوتاه (C6: 0) در Keap1-KO MEF و میتوکندری جدا شده از قلب و کبد بیشتر از میزان آنها است همتایان WT ، در حالی که در سلولهای Nrf2-KO و میتوکندری کمتر است [65]. این تأثیرات برای انسان نیز بسیار مرتبط است: در واقع ، گزارش شده است که تغییرات متابولیکی نشانگر ادغام بهتر FAO با فعالیت چرخه TCA در مطالعات مداخله انسانی با رژیم های غذایی غنی از گلوکورافانین ، پیش ماده فعال کننده کلاسیک Nrf2 سولفورافان رخ می دهد [ 66]
در طی اولین مرحله از فاو میتوکندری ، هیدروژن پرو-R برگ کربن به عنوان یک هیدرید که فاکتور FAD را به FADH2 کاهش می دهد ، که به نوبه خود الکترون ها را به ubiquinone (UbQ) در زنجیره تنفسی منتقل می کند ، در نهایت به تولید ATP کمک می کند. . در حالی که تحریک فاو توسط پالمیتوئیل کارنیتین در غیاب گلوکز باعث افزایش انتظار می رود در سطح ATP در سلولهای WT و Keap1-KO ، با افزایش ATP در سلولهای Keap1-KO سریع تر ، درمان یکسان هیچ تغییری ATP در Nrf2-KO تولید نمی کند MEFs [65]. این آزمایش نشان می دهد که ، در غیاب Nrf2 ، FAO سرکوب می شود ، و علاوه بر این ، سرکوب FAO را یکی از دلایل پایین آمدن سطح ATP در شرایط کمبود Nrf2 می داند [35] ، [64].
به طور مشخص، سلول های 293 T انسان که در آنها Nof2 خاموش شده اند، عبارتند از CPT1 و CPT2 [67]، دو ایزوفرم کارنتین پالمیتویل ترانسفراز (CPT)، آنزیم محدود کننده سرعت در FAO میتوکندری. در توافق، سطح mRNA Cpt1 در کبد Nrf2-KO در مقایسه با موش های WT [68] پایین تر است. CPT انتقال از گروه آسیل از acyl-CoA چربی بلند زنجیره ای را از کوآنزیم A به ال-کارنیتین کاتالیز می کند و در نتیجه اجازه می دهد واردات آسیکلارنیتین از سیتوپلاسم را به میتوکندری وارد کند. اگرچه تاکنون مورد بررسی قرار نگرفته است، ممکن است علاوه بر اثرات رونویسی بر روی بیان CPT1، Nrf2 همچنین ممکن است بر عملکرد این آنزیم با کنترل سطح مهارکننده آلوستریک اصلی، malonyl-CoA تاثیر بگذارد. این به این دلیل است که با مکانیزمی که در حال حاضر نامشخص است، Nrf2 منعکس کننده بیان استروییل CoA desaturase (SCD) [69] و سیترات لیائاز (CL) [69]، [70] است. جالب است که نابودی یا مهار SCD منجر به افزایش فسفریلاسیون و فعال شدن پروتئین کیناز فعال (AMPK) [71]، [72]، [73] فعال شده AMP می شود و می توان حدس زد که در غیاب Nrf2 سطح SCD افزایش می یابد، به نوبه خود کاهش فعالیت AMPK. این امر می تواند بیشتر از میزان کاهش یافته پروتئین AMPK که در مجاری موش های Nrf2-KO مشاهده شده است [68] مشاهده شود، که در توافق نزدیک با افزایش سطح AMPK مشاهده شده است که در کیپ ها و کبد های KeP1-KD گزارش شده است موشها [74]. یک نتیجه از کاهش فعالیت AMPK، تسکین آن از فسفوریلاسیون مهار کننده (در Ser79) استیل کروکوکسیلاز (ACC) [75] است که در غیاب Nrf2 می تواند بیشتر رونویسی شود زیرا آن را تحت تاثیر فعال سازی Nrf2 [70 ] فعالیت ACC بالا، در ترکیب با بیان CL اصلاح شده که تولید استیل کوا را افزایش می دهد، بستر برای ACC، در نهایت می تواند سطح محصولات ACC، malonyl-CoA را افزایش دهد. سطح بالایی از مالونیل CoA باعث جلوگیری از CPT می شود، بنابراین انتقال اسیدهای چرب به میتوکندری کاهش می یابد. در نهایت، Nrf2 به طور مثبت بیان CD36 [76]، انتقال دهنده ای است که اسیدهای چرب را در غشاهای پلاسما و میتوکندری وارد می کند. بنابراین، یک مکانیزم که توسط آن Nof2 ممکن است بر کارایی FAO میتوکندریا تأثیر بگذارد، تنظیم واردات اسیدهای چرب زنجیره طولانی به میتوکندری است.
علاوه بر مقررات رونویسی مستقیم، Nrf2 همچنین می تواند کارایی FAO میتوکندری را با تأثیرات آن بر متابولیسم مجدد سلول های سرطانی تغییر دهد. این ممکن است به خصوص هنگامی که فعالیت Nof2 کم و یا غایب باشد، شرایطی است که تغییر وضعیت سلولهای سرطانی را نسبت به حالت اکسیداسیون تغییر می دهد. در واقع، چندين آنزيم FAO به عنوان حساسيت به تغييرات بازيافت مجدد شناخته شده اند. یکی از این آنزیم، آلییل CoA dehydrogenase بسیار طولانی است (VLCAD)، که بیش از 80٪ به فعالیت dehydrogenating palmitoyl-CoA در بافت های انسانی [77] می دهد. جالب توجه است که هرد و همکاران [78] نشان داده است که VLCAD حاوی مخازن سستئین است که به طور قابل ملاحظه ای از حالت ردوکس خود پس از قرار گرفتن در معرض میتوکندری سرطان جدا شده از موش به H2O2 تغییر می کند. علاوه بر این، S-nitrosilation از VLCAD کبدی مغز در Cys238 بهبود بازده کاتالیزوری آنزیم [79] را بهبود می بخشد و احتمال دارد که اکسیداسیون همان سیستئین ممکن است اثر متفاوتی داشته باشد و در نهایت باعث کاهش کارآیی FAO میتوکندری شود. بنابراین ممکن است که اگر چه سطح بیان VLCAD در WT، Nrf2-KO یا Keap1-KO MEFs [65] به طور قابل توجهی متفاوت نیست، در صورت عدم وجود Nrf2، به دلیل سطوح بالاتر، فعالیت آنزیمی VLCAD می تواند پایین تر باشد از ROS
بر اساس همه این یافته ها ، می توان پیشنهاد کرد که (شکل 3): در صورت عدم وجود Nrf2 ، به دلیل کاهش بیان ME1 ، IDH1 ، G6PD و PGD ، سطح NADPH پایین تر است. سطح گلوتاتیون کاهش یافته نیز به دلیل کاهش بیان آنزیم هایی که در بیوسنتز و بازسازی آن شرکت می کنند و سطوح پایین تر NADPH که برای تبدیل اکسید شده به فرم کاهش یافته گلوتاتیون مورد نیاز است ، کمتر است. بیان کم ME1 باعث کاهش استخوان پیروات در ورود به میتوکندری می شود ، و گلیکولیز به منبع اصلی پیروات تبدیل می شود. تولید NADH کندتر است و منجر به اختلال در فعالیت کمپلکس I و افزایش تولید ROS میتوکندری می شود. کاهش FAD به FADH2 نیز کندتر است ، حداقل در بخشی به دلیل اکسیداسیون اسیدهای چرب با کارایی کمتر ، به خطر انداختن جریان الکترون از FADH2 به UbQ و به کمپلکس III. از آنجا که UbQH2 فعال کننده سوکسینات دهیدروژناز است [80] ، کاهش سرعت تشکیل آن ممکن است باعث کاهش فعالیت آنزیم سوکسینات دهیدروژناز شود. افزایش سطح سوپراکسید و پراکسید هیدروژن می تواند فعالیت پیچیده II را بیشتر مهار کند [81]. بازده پایین اکسیداسیون اسیدهای چرب به کاهش در دسترس بودن بستر برای تنفس میتوکندری و تولید ATP در فسفوریلاسیون اکسیداتیو کمک می کند. به عنوان یک مکانیسم جبرانی ، گلیکولیز افزایش می یابد. توابع سنتاز ATP برعکس ، به عنوان ATPase ، در تلاش برای حفظ ؟؟ متر است.
Nrf2 و بیوژنز میتوکندری
گزارش شده است که ، در مقایسه با WT ، کبد موش Nrf2-KO دارای محتوای میتوکندری پایین تر است (همانطور که توسط نسبت میتوکندری به DNA هسته تعیین می شود). این بیشتر با سرعت 24 ساعته در موش WT و Nrf2-KO کاهش می یابد. در مقابل ، اگرچه در شرایط طبیعی تغذیه ای با WT تفاوتی ندارد ، اما میزان ناشتایی در موشهایی که فعالیت Nrf2 بالایی دارند تحت تأثیر قرار نمی گیرد [82]. جالب است که مکمل با فعال کننده Nrf2 (R) -؟ - اسید لیپوئیک [83] ، [84] ، [85] باعث ایجاد پیدایش میتوکندری در سلولهای چربی 3T3-L1 می شود [86]. دو کلاس از تنظیم کننده های رونویسی هسته ای نقش مهمی در پیدایش میتوکندری دارند. طبقه اول فاکتورهای رونویسی هستند ، مانند فاکتورهای تنفسی هسته ای 11 و 2 ، که بیان ژن های رمزگذار زیرواحد پنج مجتمع تنفسی ، اجزای ترجمه میتوکندری و آنزیم های بیوسنتز هم را کنترل می کنند که در ماتریس میتوکندری قرار دارند [88]. پیانتادوسی و همکاران [89] نشان داده است که تنظیم مجدد رونویسی وابسته به Nrf2 فاکتور تنفسی هسته ای 1 باعث ایجاد بیوژنز میتوکندری می شود و از سمیت سلولی عامل شیمی درمانی آنترا سایکلین کاردوتوکسیک دوکسوروبیسین محافظت می کند. در مقابل ، ژانگ و همکاران [82] گزارش داده اند که فعال شدن ژنتیکی Nrf2 بر بیان mRNA پایه فاکتور تنفسی هسته ای 1 در کبد موش تأثیر نمی گذارد.
دسته دوم تنظیم کننده های رونویسی هسته ای با عملکردهای حیاتی در بیوژنز میتوکندری ، لخته کننده های رونویسی ، مانند گیرنده فعال شده با تکثیر پراکسی زوم هستند؟ Coactivators (PGC) 1؟ و 1؟ ، که با فاکتورهای رونویسی ، دستگاه اصلی رونویسی و RNA-splicing و آنزیم های اصلاح کننده هیستون تعامل دارند [88] ، [90] ، [91]. بیان خانواده انسدادگرهای PGC1 تحت تأثیر سیگنالهای محیطی متعددی قرار دارد. درمان فیبروبلاست های انسانی با فعال Nrf2 سولفورافان باعث افزایش توده میتوکندری و القای PGC1 می شود؟ و PGC1؟ [92] ، اگرچه وابستگی بالقوه به Nrf2 در این مطالعه مورد بررسی قرار نگرفت. با این حال ، موشهای دیابتی که Nrf2 در آنها یا توسط knapdown hypomorphic ژن Keap1 فعال می شود (db / db: Keap1flox / ؟: Nrf2 + / +) یا مختل می شوند (db / db: Keap1flox / ؟: Nrf2؟ /؟) دارای کبد PGC1 پایین تر هستند؟ سطح بیان نسبت به حیوانات کنترل (db / db: Keap1flox / +: Nrf2 + / +) [93]. هیچ تفاوتی در سطح mRNA برای PGC1 وجود ندارد؟ در کبد موشهای غیر دیابتی که WT یا Nrf2-KO هستند دیده می شود ، در حالی که این میزان در حیواناتی که بیش از حد بیانگر Nrf2 هستند (Keap1-KD و Keap1-KO مخصوص کبد) کمتر است [82]. قابل توجه است ، سرعت 24 ساعته سطح PGC1 را افزایش می دهد؟ mRNA در کبد موش ها از همه ژنوتیپ ها ، اما این افزایش در کبد Nrf2-KO در مقایسه با موش های بیان بیش از حد WT یا Nrf2 به طور قابل توجهی بیشتر است. در مقایسه با WT ، موش های Nrf2-KO که به دلیل عفونت دچار عفونت سپتیک یا آسیب حاد ریه شده اند ، تنظیم ضعیف رونویسی ضریب تنفسی هسته ای 1 و PGC1 را نشان می دهند؟ [94] ، [95]. با هم ، این مشاهدات نشان می دهد که نقش Nrf2 در حفظ سطح هر دو عامل تنفسی هسته ای و PGC1؟ پیچیده است و تحت شرایط استرس برجسته می شود.
علاوه بر بیان ژن هایی که پروتئین های میتوکندری را رمزگذاری می کنند ، بیوژنز میتوکندری نیاز به سنتز نوکلئوتیدها دارد. فعال سازی ژنتیکی Nrf2 با تنظیم مجدد مسیر پنتوز فسفات و متابولیسم فولات و گلوتامین ، به ویژه در سلول های در حال تکثیر سریع ، بیوسنتز پورین را افزایش می دهد (شکل 2) [24]. تجزیه و تحلیل رونویسی از فروخوردگی جهش یافته برای سرین میتوکندری / ترئونین کیناز پروتئین کیناز ناشی از PTEN ناشی از کیناز 1 (PINK1) نشان داده است که اختلال عملکرد میتوکندری منجر به تنظیم مجدد رونویسی ژن های متابولیسم نوکلئوتید می شود [96] ، که نشان می دهد بیوسنتز نوکلئوتید افزایش یافته است. مکانیسم محافظت در برابر پیامدهای عصبی ناشی از کمبود PINK1 را نشان می دهد. Nrf2 بیان آمیدوترانسفراز فسفریبوسیل پیرو فسفات (PPAT) را تنظیم می کند ، که ورود به مسیر بیوسنتز نوکلئوتید پورین de novo و متیلن تتراهیدروفولات دهیدروژناز 2 میتوکندری (MTHFD2) را کاتالیز می کند (شکل 2). دومی یک آنزیم دو کاره با فعالیت دهیدروژناز و سیکلوهیدرولاز است که در تأمین گلیسین و فرمات به عنوان منابع واحدهای تک کربنی برای بیوسنتز پورین در سلولهای در حال رشد بسیار حیاتی است [97]. بنابراین احتمال دارد که فعال سازی Nrf2 محافظ باشد و ممکن است نقص عملکرد میتوکندری را در کمبود PINK1 معکوس کند. در واقع ، فعال سازی دارویی Nrf2 توسط سولفورافان ، یا تری ترپنوئید RTA-408 ، بازسازی می کند و سلولهای کمبود PINK1 را در برابر سمیت دوپامین محافظت می کند [98]. اگرچه مکانیسم های اساسی پیچیده به نظر می رسند ، با هم ، این یافته ها نشان می دهد که فعالیت Nrf2 ممکن است با تأثیرگذاری بر میزان بیان فاکتورهای رونویسی مهم و لخته سازها و همچنین با افزایش بیوسنتز نوکلئوتید ، بر بیوژنز میتوکندری تأثیر بگذارد.
Nrf2 و یکپارچگی میتوکندری
اگر چه شواهد مستقیم همیشه در دسترس نیستند، نشانه های قوی وجود دارد که Nrf2 برای یکپارچگی میتوکندری ها مهم است، به خصوص در شرایط استرس اکسیداتیو. میتوکندریا جدا شده از مغز و کبد موش هایی که یک دوز مجاز از فعال کننده Nrf2 سولفورفان را تجویز کرده اند، مقاوم به باز شدن نفوذ گذار نفوذی میتوکندری (mPTP) ناشی از ترشحات بوتیل هیدروپراکسید اکسیدان [99]، [100] است. mPTP، پیچیده ای است که اجازه می دهد تا غشای داخلی میتوکندری به مولکول های قابل نفوذ با توده ها تا 1500 Da تبدیل شود، اخیرا از دیمر سنتاز F0F1-ATP [101] شکل گرفته است. مقاومت متقابل سولفورفان به باز کردن mPTP با افزایش تولید آنتی اکسیدان ارتباط دارد و سطح GSH، گلوتاتیون پراکسیداز 1، آنزیم مالیک 3 و تیرورودوکسین 2 در تمام سطوح میتوکندری جدا شده از حیوانات تحت درمان با سولفورفان [100] تنظیم می شود.
آسیب پروتئین میتوکندری و اختلال در تنفس ناشی از محصول پراکسیداسیون لیپیدهای الکتروفیلی 4-هیدروکسی-2-نننال در میتوکندری جدا شده از قشر مغز موشهای تحت درمان با سولفورافان ضعیف می شود [102]. در سلولهای اپیتلیال کلیوی موش و در کلیه ، سولفورافان در برابر سمیت ناشی از سیس پلاتین و جنتامایسین و از دست دادن ؟؟ متر محافظت می کند [103] ، [104]. محافظت در برابر پانل اکسیدان ها (سوپراکسید ، پراکسید هیدروژن ، پراکسینیتریت) و الکتروفیل ها (4-هیدروکسی-2-نننال و آکرولین) و افزایش دفاع آنتی اکسیدانی میتوکندری نیز در هنگام درمان سلول های عضله صاف آئورت موش با سولفورافان مشاهده شده است [105 ] در مدلی از آسیب حاد کلیوی ناشی از کنتراست ، اخیراً نشان داده شده است که پیش شرطی سازی ایسکمیک اندام دارای اثرات محافظتی ، از جمله مهار باز شدن mPTP و تورم میتوکندری ، با فعال شدن Nrf2 متعاقب مهار GSK3 است؟ [106]
میتوفاژی ، فرایندی که میتوکندری های ناکارآمد به طور انتخابی توسط اتوفاگوزوم ها فرو رفته و به لیزوزوم ها تحویل داده می شوند تا توسط سلول تخریب و بازیافت شوند ، برای هموستاز میتوکندری ضروری است [107] ، [108]. در حالی که هیچ رابطه علتی بین Nrf2 و میتوفاژی ایجاد نشده است ، شواهدی وجود دارد که فاکتور رونویسی با بازی در میتوفاژی ممکن است در کنترل کیفیت میتوکندری مهم باشد. این ممکن است به ویژه در شرایط استرس اکسیداتیو برجسته باشد. بنابراین ، در یک مدل از سپسیس ، افزایش سطح نشانگر اتوفاگوزوم MAP1 زنجیره سبک 3-II (LC3-II) و پروتئین باری p62 در 24 ساعت پس از عفونت در Nrf2-KO در مقایسه با موش WT سرکوب می شود [109] . اخیراً یک مولکول کوچک القا کننده میتوفاژی (به نام القا mit کننده میتوفاژی با واسطه P62). این ترکیب 1,4،1,2,3-دی فنیل-2،1،110-تریازول در ابتدا به عنوان یک فعال کننده Nrf2 طراحی شده است که برهمکنش فاکتور رونویسی با Keap1 را مختل می کند [1]. مشابه سلول هایی که در آنها Nrf3 از نظر ژنتیکی تنظیم مجدد شده است (Keap2-KD یا Keap2-KO) ، سلول های در معرض PMI دارای استراحت بالاتری هستند؟ نکته مهم ، افزایش محلی سازی LCXNUMX در میتوکندری است که پس از درمان PMI سلول های WT مشاهده می شود ، در سلول های NrfXNUMX-KO رخ نمی دهد ، که نشانگر درگیری NrfXNUMX است.
اخیرا، تجزیه و تحلیل فراوانی کبد، وجود میتوکندری متورم با کاهش کریستا و غشاهای متفرع شده در hepatocytes از Nrf2-KO، اما نه WT، موش هایی که از یک رژیم غذایی با چربی بالا برای هفته های 24 تغذیه می کردند؛ به ویژه، این لیپدها شواهد واضحی از استرس اکسیداتیو و التهاب [68] را نشان می دهد. می توان نتیجه گرفت که Nrf2 نقش مهمی در حفظ یکپارچگی میتوکندری در شرایط استرس اکسیداتیو و التهابی دارد.
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
Nrf2 فاکتور رونویسی است که نقش مهمی در سیستم دفاع ضد عفونی سلولی بدن انسان ایفا می کند. عنصر پاسخ دهنده آنتی اکسیدان، یا ARE، مکانیسم قانونی ژن است. بسیاری از مطالعات انجام شده نشان داده است که Nrf2 یا عامل NF-E2 مرتبط با 2، طیف گسترده ای از ژن های هدایت شده ARE را در چندین سلول تنظیم می کند. Nrf2 نیز نقش مهمی در حفاظت سلولی و ضد سرطان زا داشت، که نشان می دهد که Nrf2 ممکن است یک درمان مؤثر در مدیریت بیماری های نورودنژراتیک و سرطان هایی است که به نظر می رسد ناشی از استرس اکسیداتیو است. دکتر الکس جیمنز DC، CCST Insight
اظهارات نهایی
اگر چه بسیاری از سوالات هنوز هم باز هستند، شواهد تجربی موجود به وضوح نشان می دهد که Nrf2 یک بازیکن مهم در حفظ هومیوستاز میتوکندری و یکپارچگی ساختاری است. این نقش به ویژه در شرایط استرس اکسیداتیو، الکتروفیلی و التهابی نقش مهمی ایفا می کند زمانی که توانایی بالا بردن پاسخ های محافظتی محافظت شده توسط Nrf2 بر سلامت کلی و بقا سلول و ارگانیسم تاثیر می گذارد. نقش Nrf2 در عملکرد میتوکندری نشان دهنده لایه دیگری از مکانیسم های سیتوپروتئینی وسیع است که توسط این عامل رونویسی سازگار شده است. همانطور که بسیاری از شرایط بیماری های انسانی، استرس اکسیداتیو، التهاب و اختلال عملکرد میتوکندری را بعنوان اجزای ضروری پاتوژنز می دانند، فعال سازی فارماکولوژیک Nrf2، وعده ای برای پیشگیری و درمان بیماری ها است. درک جامع از مکانیسم دقیق که توسط Nof2 اثر میتوکندری تاثیر می گذارد برای طراحی منطقی آزمایش های بالینی آینده ضروری است و ممکن است نشانگرهای جدید برای نظارت بر اثربخشی درمان باشد.
هدف مقاله بالا بحث و همچنین نشان دادن نقش نوظهور Nrf2 در عملکرد میتوکندری بود. Nrf2 ، یا عامل فاکتور هسته ای erythroid 2، یک تنظیم کننده در حال ظهور مقاومت سلولی در برابر اکسیدان ها است که می تواند به استرس اکسیداتیو کمک کند ، بر عملکرد سلول تأثیر می گذارد و منجر به ایجاد سمیت ، بیماری مزمن و حتی سرطان می شود. در حالی که تولید اکسیدان در بدن انسان می تواند اهداف مختلفی را شامل شود ، از جمله تقسیم سلولی ، التهاب ، عملکرد ایمنی ، اتوفاژی و پاسخ استرس ، کنترل تولید بیش از حد آنها برای جلوگیری از مسائل بهداشتی ضروری است. دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
درد پشتاین یکی از مهمترین دلایل معلولیت و روزهای از دست رفته در کار در سراسر جهان است. کمردرد دومین دلیل شایع مراجعه به مطب توسط پزشکان است که بیشتر از عفونت های دستگاه تنفسی فوقانی است. تقریباً 80 درصد مردم حداقل یک بار در طول زندگی خود درد کمر را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده است که از استخوان ها ، مفاصل ، رباط ها و ماهیچه ها در میان سایر بافت های نرم تشکیل شده است. به همین دلیل ، صدمات و یا شرایط وخیم مانند دیسک های فتق دیسک، می تواند در نهایت به علائم کمردرد منجر شود. آسیب های ورزشی یا صدمات ناشی از تصادفات اتومبیل اغلب شایعترین علت کمر درد است ، با این حال ، گاهی اوقات ساده ترین حرکات می تواند نتایج دردناکی داشته باشد. خوشبختانه ، گزینه های درمانی جایگزین مانند مراقبت های کایروپراکتیک ، می توانند با استفاده از تنظیمات ستون فقرات و دستکاری های دستی ، درد کمر را کاهش دهند و در نهایت تسکین درد را بهبود بخشند.
Nrf2 از فعال شدن گروهی از آنزیم های آنتی اکسیدان و سم زدایی آن ها و ژن هایی که بدن انسان را از اثرات مسائل مربوط به سلامت مرتبط با افزایش سطوح استرس اکسیداتیو مانند بیماری آلزایمر محافظت می کند، حمایت می کند. انواع مختلفی از مواد طبیعی برای فعال کردن مسیر Nrf2 نشان داده شده است که می تواند به نشانه های بیماری های نورودنراتیو کمک کند. هدف از مقاله زیر این است که در مورد نقش اساسی Nrf2 ناشی از التهاب مزمن بحث شود.
چکیده
التهاب شایعترین ویژگی بسیاری از بیماریها و عوارض مزمن است ، در حالی که نقش مهمی در سرطان زایی دارد. چندین مطالعه نشان داده است که Nrf2 با هماهنگی در جذب سلولهای التهابی و تنظیم بیان ژن از طریق عنصر پاسخ آنتی اکسیدانی (ARE) به روند ضد التهابی کمک می کند. مسیر سیگنالینگ Keap1 (پروتئین مرتبط با ECH مرتبط با Kelch) / Nrf2 (عامل 2 مربوط به NF-E45 p2) / ARE عمدتا بیان ژن ضد التهاب را تنظیم می کند و از پیشرفت التهاب جلوگیری می کند. بنابراین ، شناسایی مواد شیمیایی شیمیایی ضدالتهابی وابسته به Nrf2 به یک نکته کلیدی در کشف دارو تبدیل شده است. در این بررسی ، ما در مورد اعضای مسیر سیگنال Keap1 / Nrf2 / ARE و ژن های پایین دست آن ، اثرات این مسیر بر روی مدل های حیوانی بیماری های التهابی و پیوند متقاطع با مسیر NF-؟ B بحث می کنیم. علاوه بر این ، ما همچنین در مورد تنظیم التهاب NLRP3 توسط Nrf2 بحث می کنیم. علاوه بر این ، ما سناریوی فعلی تولید مواد شیمیایی شیمیایی ضد التهابی و موارد دیگر را که مسیر سیگنالینگ Nrf2 / ARE را واسطه قرار می دهند ، خلاصه می کنیم.
التهاب یک فرایند پیچیده است که وقتی که بافت ها توسط محرک های مضر نظیر پاتوژن، آسیب یا تحریک کننده آلوده یا زخمی می شوند، رخ می دهد. سلول های ایمنی، رگ های خونی و واسطه های مولکولی در این واکنش محافظتی [1] دخیل هستند. التهاب نیز یک پدیده پاتولوژیک است که با انواع بیماری های بیماری ای همراه است که به طور عمده توسط عوامل فیزیکی، شیمیایی، بیولوژیکی و روانی ایجاد می شود. هدف التهاب، محدود کردن و از بین بردن علل آسیب سلولی، پاک کردن و / یا جذب سلول های بنیادی و بافت های نئروتیک است و تعمیرات بافت را آغاز می کنند. دو فرم متمایز التهاب مشخص است: حاد و مزمن. التهاب حاد خود محدود کننده است و برای میزبان سودمند است، اما التهاب مزمن مزمن یکی از ویژگی های رایج بسیاری از بیماری ها و عوارض مزمن است. نفوذ مستقیم توسط بسیاری از سلول های ایمنی تک هسته ای مانند مونوسیت ها، ماکروفاژها، لنفوسیت ها و سلول های پلاسما، و همچنین تولید سیتوکین های التهابی باعث التهاب مزمن می شود. به رسمیت شناخته شده است که التهاب مزمن نقش مهمی در سرطانزا دارد [2]. به طور کلی، هر دو مسیر سیگنالینگ پروتئین و ضد التهابی در فرآیند التهابی نرمال تعامل دارند.
در فرآیند التهابی پاتولوژیک ، سلولهای بنیادی ، مونوسیت ها ، ماکروفاژها ، لنفوسیت ها و سایر سلول های ایمنی ابتدا فعال می شوند. سپس سلول ها به محل آسیب دیدگی جذب می شوند و در نتیجه تولید گونه های اکسیژن واکنش پذیر (ROS) ایجاد می شود که به مولکول های ماکرو از جمله DNA آسیب می رساند. همزمان ، این سلولهای التهابی مقادیر زیادی واسطه التهابی مانند سیتوکین ، کموکین و پروستاگلاندین را نیز تولید می کنند. این واسطه ها بیشتر ماکروفاژها را در مکان های موضعی التهاب استخدام می کنند و آبشارهای انتقال سیگنال و فاکتورهای رونویسی مرتبط با التهاب را مستقیماً فعال می کنند. مسیرهای سیگنالینگ NF-؟ B (فاکتور هسته ای کاپا B) ، MAPK (پروتئین کیناز فعال شده با میتوژن) و JAK (ژانوس کیناز) -STAT (مبدل سیگنال و فعال کننده رونویسی) در توسعه مسیر کلاسیک التهاب نقش دارند. [3] ، [4] ، [5] مطالعات قبلی نشان داده است که فاکتور رونویسی Nrf2 (عامل 2 مربوط به NF-E45 p2) بیان آنزیم های سم زدایی فاز II از جمله NADPH ، NAD (P) H quinone oxidoreductase 1 ، گلوتاتیون پراکسیداز ، فریتین ، هم اکسیژناز -1 (HO) را تنظیم می کند. -1) ، و ژن های آنتی اکسیدان که از طریق اثرات ضد التهابی سلول ها را از آسیب های مختلف محافظت می کنند ، بنابراین بر روند بیماری تأثیر می گذارند [6] ، [7] ، [8].
با توجه به این یافته های قابل توجه، توسعه داروهای هدفمند برای بیماری های التهابی از طریق مسیرهای سیگنالینگ، در سال های اخیر، علاقه فراوانی به خود جلب کرده است. در این بررسی، تحقیقات در مورد سیگنالینگ التهاب در Keap1 (پروتئین وابسته به ECH کلچ) / Nrf2 (NF-E2 p45 مرتبط با عامل 2) / ARE (عنصر پاسخ دهنده آنتی اکسیدانی) خلاصه می شود.
ساختار و مقررات Nrf2
مقررات Nrf1 وابسته به Keap2-dependent
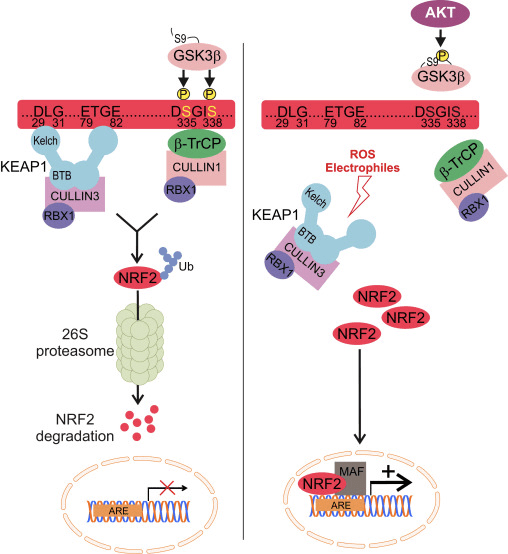
Nrf2 به زیرخانواده Cap n Collar (CNC) تعلق دارد و شامل هفت حوزه عملکردی ، Neh (همسانی Nrf2-ECH) 1 تا Neh7 [9] ، [10] است. Neh1 یک دامنه CNC-bZIP است که به Nrf2 اجازه می دهد تا با پروتئین ، DNA و سایر شرکای رونویسی عضلانی ، فیبروسارکومای عضلانی-آپونوروتیک (Maf) دگرگونی یابد و همچنین با آنزیم متصل کننده یوبی کویتین UbcM2 [11] ، [12] یک مجتمع هسته ای تشکیل دهد. Neh2 حاوی دو نقش مهم است که به عنوان DLG و ETGE شناخته می شوند ، که برای تعامل بین Nrf2 و تنظیم کننده منفی آن Keap1 ضروری است [13] ، [14].
Keap1 یک آداپتور پایه ای برای لیزوز EXINUMX ubiquitin لیگاز مبتنی بر Cullin است، که از طریق فعالیت ubiquitination و تخریب پروتئازام در شرایط عادی [3]، [2]، [15]، فعالیت های رونویسی Nof16 را مهار می کند. دامنه KELCH از homodimer Keap17 با DLG و ETGE از دامنه Nrf1-Neh2 در سیتوزول، جایی که ETGE عمل می کند به عنوان یک لولا با بالا بودن وابستگی و DLG به عنوان یک لنگ [2] عمل می کند. تحت استرس اکسیداتیو یا تحت تاثیر قرار گرفتن با activators Nrf18، Nrf2 از اتصال Keap2 به علت اصلاح تیئول از کپسول های سیتین Keap1، که در نهایت از ubiquitination Nrf1 و تخریب پروتئازOMAL [2] جلوگیری می کند، از هم جدا می شود. سپس Nrf19 به هسته منتقل می شود، با پروتئین های کوچک Maf ها heterodimerizes، و باتری ژن ARE (شکل 2A) را فعال می کند. Terminal Carboxy از Neh1 به عنوان یک دامنه ترانسکتیویته با تعامل با CO-activator شناخته شده به عنوان CHD3 (پروتئین اتصال DNA chromo-ATPase / helicase DNA) [6] عمل می کند. Neh20 و Neh4 نیز به عنوان دامنه های transactivation عمل می کنند، اما پیوند دیگری با همکاری سازنده رونویسی شناخته شده به نام CBP (پروتئین متصل کننده پروتئین cAMP-response-binding) [5] دارند. علاوه بر این، Neh21 و Neh4 با COFACTOR RAC5 / AIB3 / SRC-1 همکاری می کنند، که منجر به افزایش بیان ژن ARE [xnumx] شده با Nrf3 می شود. Neh2 دارای یک سیگنال صادراتی هسته ای حساس به حساس به نور است که برای تنظیم و مکان سازی سلول Nrf22 [5] بسیار مهم است.
شکل 1 Keap1 وابسته و مستقل تنظیم Nrf2. (A) تحت شرایط پایه ، Nrf2 توسط Keap1 توسط دو نقوش (ETGE و DLG) که منجر به Ubiquitination با واسطه CUL3 و به دنبال آن تخریب پروتئازوم می شود ، جدا می شود. تحت استرس اکسیداتیو ، Nrf2 از Keap1 جدا می شود ، به هسته منتقل می شود و باتری ژن ARE را فعال می کند. (B) GSK3 فسفریله Nrf2 می کند و این شناخت Nrf2 توسط؟ -TrCP را برای ubiquitination با واسطه CUL1 و تخریب پروتئازوم بعدی تسهیل می کند. (C) p62 با Keap1 جداسازی می شود و منجر به تخریب اتوفاژیک آن ، آزادسازی Nrf2 و افزایش سیگنالینگ Nrf2 می شود.
Keap1 مستقل Nrf2 مقررات
شواهد در حال ظهور مکانیسم جدیدی از تنظیم Nrf2 را نشان می دهد که مستقل از Keap1 است. حوزه Neh6 غنی از سرین Nrf2 با اتصال با دو نقش خود (DSGIS و DSAPGS) به پروتئین حاوی تکرار α ترانسدوین (؟ -TrCP) نقش مهمی در این تنظیم دارد. ؟ -TrCP یک گیرنده سوبسترا برای کمپلکس لیپاز یوبی کویتین Skp24 Cul1 Rbx1 / Roc1 است که Nrf1 را برای ubiquitination و تخریب پروتئازومی هدف قرار می دهد. گلیکوژن سنتاز کیناز -2 یک پروتئین حیاتی است که در تثبیت و تنظیم Nrf3 مستقل از Keap1 نقش دارد. آن را برای تسهیل تشخیص Nrf2 توسط؟ -TrCP و تخریب پروتئین بعدی ، Nrf2 را در حوزه Neh6 فسفریله می کند (شکل 2 B).
دیگر تنظیم کننده Nrf2
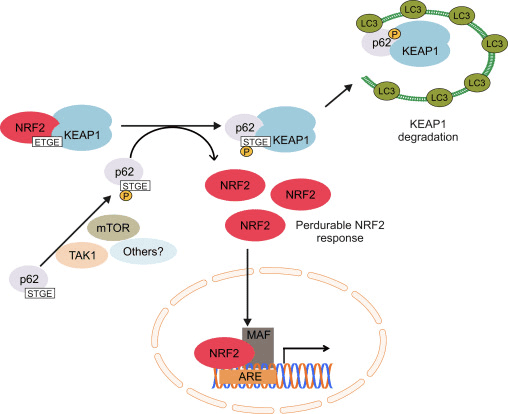
یکی دیگر از شواهد نشان داده است یک مسیر غیر متعارف از فعال شدن p62 وابسته Nrf2 که در آن p62 sequesters Keap1 به تخریب اتوفاژی که در نهایت به ثبات Nrf2 و transactivation وابسته به Nrf2 ژن [26] منجر می شود، [27]، [ 28]، [29] (شکل 1C).
شواهد جمع آوری شده نشان می دهد که چندین miRNA نقش مهمی در تنظیم فعالیت Nrf2 دارند [30]. سانگوکويا و همکاران [31] نشان داد که miR-144 به طور مستقیم فعالیت Nrf2 را در رده سلولی K562 لنفوبلاست ، سلولهای اولیه اریتروئید انسانی انسان و رتیکولوسیت های بیماری سلول داسی کاهش می دهد. یک مطالعه جالب دیگر در سلولهای اپیتلیال پستان انسان نشان داد که miR-28 از طریق مکانیزمی مستقل از Keap2 باعث مهار Nrf1 می شود [32]. به همین ترتیب ، miRNA های miR-153 ، miR-27a ، miR-142-5p و miR144 بیان Nrf2 را در رده سلولی SH-SY5Y عصبی کاهش می دهند [33]. سینگ و همکاران [34] نشان داد که بیان خارج رحمی miR-93 بیان ژن های تنظیم شده با Nrf2 را در یک مدل موش صحرایی ناشی از سرطان پستان 17؟ استرادیول (E2) کاهش می دهد.
یک کشف اخیر از آزمایشگاه ما یک بازدارنده درون زا از Nrf2 شناخته شده به عنوان گیرنده X رتینوئیک آلفا (RXR؟) شناخته شده است. RXR؟ یک گیرنده هسته ای است ، از طریق حوزه اتصال DNA (DBD) با حوزه Neh7 Nrf2 (باقیمانده اسید آمینه) ارتباط برقرار می کند و به طور خاص فعالیت Nrf209 را در هسته مهار می کند. علاوه بر این ، گیرنده های هسته ای دیگر مانند گیرنده های فعال شده با پرولیسایر پراكسیزوم- ، ER؟ ، گیرنده های مربوط به استروژن- و گیرنده های گلوكوكورتیكوئید نیز گزارش شده اند كه مهاركننده های درون زایی فعالیت Nrf316 هستند [2] ، [2].
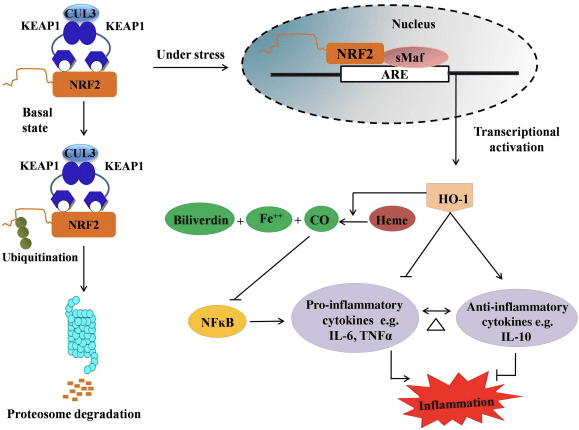
نقش ضد التهابی محور Nrf2 / HO-1
HO-1 ایزوفرم القایی و آنزیم محدود کننده سرعت است که تجزیه هم به مونوکسید کربن (CO) و آهن آزاد و بیلیفردین را به بیلی روبین تبدیل می کند. تخریب آنزیمی فهم آزاد الکل و همچنین تولید ترکیبات ضد التهابی مانند CO و بیلی روبین نقش مهمی در حفظ اثرات محافظتی HO-1 (شکل 2) دارد.
شکل 2 خلاصه مسیر Nof2 / HO-1. تحت شرایط پایه ، Nrf2 به سرکوبگر Keap1 متصل می شود که منجر به ubiquitination و به دنبال آن تخریب پروتئازوم می شود. در طول استرس اکسیداتیو ، Nrf2 آزاد به هسته جابجا می شود ، در آنجا با اعضای خانواده کوچک ماف کم می شود و به ژن های ARE مانند HO-1 متصل می شود. HO-1 تنظیم شده ، هم را به CO ، بیلی روبین و آهن آزاد تبدیل می کند. CO به عنوان یک بازدارنده از مسیر NF-؟ B عمل می کند که منجر به کاهش بیان سیتوکین های پیش التهاب می شود ، در حالی که بیلی روبین نیز به عنوان آنتی اکسیدان عمل می کند. علاوه بر این ، HO-1 به طور مستقیم باعث مهار سیتوکین های پیش التهابی و همچنین فعال سازی سیتوکین های ضد التهاب می شود ، بنابراین منجر به تعادل فرآیند التهابی می شود.
Nrf2 با افزایش بیان mRNA و پروتئین ژن HO-1 را القا می کند و این یکی از ژن های کلاسیک تنظیم شده با Nrf2 است که به طور گسترده ای در مطالعات in vitro و in vivo استفاده می شود. چندین مطالعه نشان داده است که HO-1 و متابولیت های آن اثرات ضد التهابی قابل توجهی با واسطه Nrf2 دارند. افزایش بیان HO-1 که با Nrf2 فعال انجام می شود منجر به مهار نتایج سیگنالینگ NF؟ B در کاهش آسیب مخاط روده و اختلال عملکرد اتصال تنگ در مدل پیوند کبد موش صحرایی نر Sprague-Dawley می شود [35]. تنظیم بالای بیان HO-2 وابسته به Nrf1 ممکن است از میوبلاست C2C12 مشتق شده از موش در برابر سمیت سلولی H2O2 محافظت کند [36]. HO-2 وابسته به Nrf1 بر پاسخهای التهابی ناشی از لیپوپلی ساکارید (LPS) در RAW264.7- یا ماکروفاژهای سلول کف کف ماکروفاژ صفاقی موش تأثیر دارد. فعالیت Nrf2 فنوتیپ ماکروفاژهای سلول فوم حساس به حساسیت و جلوگیری از التهاب بی حد و حصر ماکروفاژها ، آنها نقش مهمی در پیشرفت تصلب شرایین دارند [37]. محور Nrf2 / HO-1 سلولهای میکروگلیایی موش BV2 ناشی از LPS و سلولهای HT22 هیپوکامپ موش را تحت تأثیر قرار می دهد و بر التهاب عصبی تأثیر می گذارد. تنظیم مجدد بیان HO-1 از طریق مسیر Nrf2 در سلول های میکرولیلی BV2 موش که از مرگ سلولی سلول های HT22 هیپوکامپ موش دفاع می کنند [38]. علاوه بر این ، مولکولهای ترکیبی مبتنی بر کبالت (HYCOs) که یک القا کننده Nrf2 را با انتشار دهنده مونوکسیدکربن (CO) ترکیب می کنند ، بیان Nrf2 / HO-1 را افزایش می دهد ، CO را آزاد می کند و در شرایط in vitro فعالیت ضد التهابی انجام می دهد. HYCO ها همچنین HO-1 بافت را تنظیم کرده و پس از تجویز داخل بدن ، CO را در خون تحویل می دهند و از استفاده بالقوه آنها در برابر شرایط التهابی پشتیبانی می کنند [39]. تنظیم مجدد Nrf2 / HO-1 با افزایش فعالیت فروسیتی ماکروفاژهای موش تحت درمان با کلرامین های تورین ، التهاب را کاهش می دهد [40]. در کل ، مدل های آزمایشی توضیح داده شده در بالا نشان داد که محور Nrf2 / HO-1 نقش اصلی در عملکرد ضد التهابی دارد ، نشان می دهد که Nrf2 یک هدف درمانی در بیماری های مرتبط با التهاب است.
علاوه بر این، فرآورده های HO-1 مانند CO، بیلی روبین، به عنوان یک آنتی اکسیدان قوی در طول استرس اکسیداتیو و آسیب سلولی [41]، [42]؛ آن را به سلولهای سرطانی و هپاتیت می رساند [43]، [44]؛ جلوگیری از تولید iNOS و NO [45]، [46]، [47] از موش ها و موش ها در برابر شوک اندوتوکسیک محافظت می کند. علاوه بر این، بیلیروبین فعال سازی اندوتلیال و اختلال عملکرد [48] را کاهش می دهد. جالب توجه است، بیلی روبین انتقال مونوکلونال leukocytes را از طریق مولکول چسبندگی 1 [49] کاهش می دهد. این منابع خاص که نشان می دهد نه تنها HO-1 به عنوان یک عامل ضد التهاب قوی بلکه متابولیت های آن عمل می کند.
واسطه های التهابی و آنزیم های مهار شده توسط Nrf2
سیتوکین ها و کومکوکان ها
سیتوکین ها پروتئین ها و پلی پپتیدهای با وزن مولکولی کم هستند که توسط سلول های مختلف ترشح می شوند. آنها رشد ، تمایز و عملکرد ایمنی سلول را تنظیم می کنند و در التهاب و ترمیم زخم نقش دارند. سیتوکین ها شامل اینترلوکین ها (IL) ، اینترفرون ها ، فاکتور نکروز تومور (TNF) ، عامل تحریک کلنی ، کموکین ها و فاکتورهای رشد هستند. برخی از سیتوکین ها به عنوان واسطه های پیش التهاب شمرده می شوند در حالی که برخی دیگر عملکرد ضد التهابی دارند. قرار گرفتن در معرض استرس اکسیداتیو منجر به تولید بیش از حد سیتوکین ها می شود که باعث استرس اکسیداتیو در سلول های هدف می شود. هنگام فعال شدن NF-؟ B توسط استرس اکسیداتیو ، چندین سیتوکین پیش التهابی بیش از حد تولید می شوند. علاوه بر این ، استرس اکسیداتیو پیش التهابی باعث فعال شدن بیشتر NF-؟ B و تولید بیش از حد سیتوکین ها می شود. فعال سازی سیستم Nrf2 / ARE نقش مهمی در برهم زدن این چرخه دارد. کموکاین ها خانواده ای از سیتوکین های کوچک هستند که نقش اصلی آنها هدایت مهاجرت سلول های التهابی است. آنها عمدتا به عنوان کشنده های شیمیایی برای لکوسیت ها ، مونوسیت ها ، نوتروفیل ها و سایر سلول های موثر عمل می کنند.
گزارش شده است که فعال شدن Nrf2 مانع از تنظیم مجدد رونویسی ناشی از LPS سیتوکین های پیش التهاب ، از جمله IL-6 و IL-1 می شود؟ [50] IL-1؟ و تولید IL-6 نیز در Nrf2؟ /؟ افزایش یافته است. موش های مبتلا به کولیت ناشی از سولفات دکستران [51] ، [52]. Nrf2 تولید IL-17 پایین دست و سایر عوامل التهابی Th1 و Th17 را مهار می کند و روند بیماری را در یک مدل آزمایشی مولتیپل اسکلروزیس ، انسفالیت خود ایمنی سرکوب می کند [53]. ژن های ضد اکسیدان وابسته به Nrf2 HO-1 ، NQO-1 ، Gclc و Gclm TNF- ؟، IL-6 ، پروتئین 1 شیمیایی جاذب مونوسیت ها (MCP1) ، پروتئین التهابی 2 ماکروفاژ (MIP2) و التهابی واسطه ها اما در مورد موشهای حذفی Nrf2 ، اثر ضد التهابی رخ نمی دهد [54]. نوتروفیل های صفاقی موش های ناک اوت Nrf2 تحت درمان با LPS دارای میزان سیتوکین (TNF-؟ و IL-6) و کموکاین (MCP1 و MIP2) به طور قابل توجهی بالاتر از سلول های نوع وحشی (WT) هستند [54]. در شرایط آزمایشگاهی ، انتقال ژن Nrf2 به سلولهای عضلانی صاف آئورت انسان و خرگوش ترشح MCP1 [8] ، [55] را سرکوب می کند و بیان HO-2 وابسته به Nrf1 سرکوب NF-؟ B و MCP-1 - تحریک شده TNF -؟ ترشح در سلولهای اندوتلیال ورید ناف انسان [56]. این یافته ها حاکی از آن است که ، در پاسخ به محرک های التهابی ، تنظیم مجدد سیگنالینگ Nrf2 مانع تولید بیش از حد سیتوکین ها و کموکاین های پیش التهابی و همچنین محدود کردن فعال شدن NF-؟ B می شود.
مولکول های چسبندگی سلولی
مولکول های چسبندگی سلولی (CAM) پروتئین هایی هستند که با سلول ها یا با ماتریکس خارج سلول همراه می شوند. آنها در سطح سلول قرار دارند و در شناسایی سلول ، فعال سازی سلول ، انتقال سیگنال ، تکثیر و تمایز نقش دارند. در میان CAM ها ، ICAM-1 و VCAM-1 از اعضای مهم خانواده فوق العاده ایمونوگلوبولین هستند. ICAM-1 در غلظت های کم در غشای سلولهای لکوسیتی و اندوتلیال وجود دارد. با تحریک سیتوکین ، غلظت به طور قابل توجهی افزایش می یابد. ICAM-1 می تواند توسط IL-1 و TNF القا شود و توسط اندوتلیوم عروقی ، ماکروفاژها و لنفوسیت ها بیان می شود. این یک لیگاند برای اینتگرین است ، یک گیرنده موجود در لکوسیت ها. هنگامی که پل ICAM-1-integrin فعال می شود ، لکوسیت ها به سلول های اندوتلیال متصل می شوند و سپس به بافت های زیر اندوتلیال مهاجرت می کنند [57]. VCAM-1 واسطه چسبندگی لنفوسیت ها ، مونوسیت ها ، ائوزینوفیل ها و بازوفیل ها به اندوتلیوم عروقی است و به استخدام لکوسیت ها کمک می کند ، که در نهایت منجر به آسیب بافتی به دلیل استرس اکسیداتیو می شود. Nrf2 فعالیت پروموتر VCAM-1 را مهار می کند [58]. ژن پایین دست تنظیم شده توسط Nrf2 HO-1 می تواند بیان E-سلکتین و VCAM-1 ، مولکول های چسبندگی مرتبط با سلول های اندوتلیال را تحت تأثیر قرار دهد [59]. بیان ریوی چندین CAM مانند CD-14 ، TREM1 ، SELE ، SELP و VCAM-1 در Nrf2؟ /؟ به طور قابل توجهی بالاتر است. موشها نسبت به موشهای Nrf2 + / + [60]. Nrf2 در سلولهای اندوتلیال آئورت انسانی بیان VCAM-1 ناشی از TNF را سرکوب می کند و در چسبندگی سلولهای monocytic U937 ناشی از TNF - β تداخل ایجاد می کند [8]. بیان بیش از حد Nrf2 همچنین باعث مهار بیان ژن VCAM-1 ناشی از TNF در سلولهای اندوتلیال ریز عروقی انسان می شود [61]. آنتی اکسیدان 3-هیدروکسی آنتاریلیک اسید (HA) که به طور طبیعی وجود دارد ، یکی از متابولیت های ال تریپتوفان است که در داخل بدن در طول مسیر متابولیک شناخته شده به عنوان مسیر کینورنین در طول التهاب یا عفونت ایجاد می شود ، بیان HO-1 را تحریک می کند و Nrf2 را در ناف انسان تحریک می کند. سلولهای اندوتلیال وریدی (HUVECs). بیان HO-2 وابسته به Nrf1 ناشی از HA ترشح MCP-1 ، بیان VCAM-1 و فعال سازی NF-kB همراه با آسیب عروقی و التهاب در تصلب شرایین را مهار می کند [56]. مشتقات کالکون مصنوعی ضد تکثیری و ضد التهابی 2؟ ، 4؟ ، 6؟ -tris (متوکسیمتوکسی) کلکون باعث مهار ICAM-1 ، سیتوکین پیش التهابی IL-1؟ و TNF-؟ می شود. بیان در بافت کولون از موش های تحت درمان با ترینیتروبنزن سولفونیک اسید [62]. تنظیم مجدد Nrf2 بیان ICAM-1 ناشی از TNF را در سلولهای اپیتلیال رنگدانه شبکیه انسان تحت درمان با لیکوپن مهار می کند [63]. تمام این مطالعات نشان می دهد که Nrf2 با تنظیم مهاجرت و نفوذ سلولهای التهابی به بافت ملتهب ، نقشی اساسی در روند التهابی ایفا می کند.
ماتریکس متالوپروتئیناز (MMPs)
MMP ها به طور گسترده در ماتریکس خارج سلولی وجود دارند و در فرایندهای فیزیولوژیکی و آسیب شناختی مانند تکثیر سلولی ، مهاجرت ، تمایز ، ترمیم زخم ، آنژیوژنز ، آپوپتوز و متاستاز تومور نقش دارند. گزارش شده است که محور Nrf2 / HO-1 باعث مهار MMP-9 در ماکروفاژها و MMP-7 در سلولهای اپیتلیال روده انسان می شود و این در درمان بیماری التهابی روده مفید است [62] ، [64]. آسیب پوستی ناشی از اشعه ماورا بنفش در ناک اوت Nrf2 شدیدتر از موش WT است و سطح MMP-9 به طور قابل توجهی بالاتر است ، نشان می دهد که Nrf2 بیان MMP-9 را کاهش می دهد. بنابراین ، Nrf2 به عنوان محافظ در برابر تابش اشعه ماورا بنفش در نظر گرفته می شود [65]. مطالعه دیگری همچنین گزارش داد که فعال سازی رونویسی تنظیم نشده MMP-9 در حمله سلول و تومور از طریق مهار مسیر سیگنالینگ NF-kB تنظیم می شود [66]. در آسیب نخاعی آسیب دیده ، مسیر سیگنالینگ NF-kB نیز در تنظیم سطح mRNA MMP-9 نقش دارد [67]. بنابراین ، در التهاب تنظیم MMPs مستقیماً از طریق مسیر Nrf2 یا غیر مستقیم از طریق مسیر NF-؟ B تحت تأثیر Nrf2 تحت تأثیر قرار می گیرد.
Cyclooxygenase-2 (COX2) و سنتز اکسید نیتریک قابل انجماد (INOS)
یک سری آزمایشات روی موشهای ناک اوت Nrf2 نقش اساسی آن را در التهاب و تنظیم ژن های پیش التهاب مانند COX-2 و iNOS نشان داده است. برای اولین بار ، خور و همکاران افزایش بیان سیتوکین های پیش التهابی مانند COX-2 و iNOS در بافت های روده بزرگ Nrf2؟ /؟ موشها با موشهای WT Nrf2 + / + مقایسه شدند ، این نشان می دهد که Nrf2 فعالیت آنها را سرکوب می کند [51]. گزارش دیگری در مورد پیش تصفیه با سولفورافان ، یکی از فعال کننده های شناخته شده Nrf2 در سبزیجات چلیپایی ، اثر ضد التهابی آن را در مهار بیان TNF- ؟، IL-1 ؟، COX-2 و iNOS در هر دو mRNA نشان داد. سطح پروتئین و پروتئین در ماکروفاژهای صفاقی اولیه از موش های Nrf2 + / + در مقایسه با کسانی که از Nrf2؟ /؟ موش [68]. به طور مشابه ، هیپوکامپ موش های Nrf2-knockout با التهاب ناشی از LPS نیز بیان بالاتر مارکرهای التهابی مانند iNOS ، IL-6 و TNF-؟ را نشان می دهد. از موشهای WT [69]. به همین ترتیب ، موش های ناک اوت Nrf2 نسبت به استرس اکسیداتیو ناشی از 1-متیل-4-فنیل-1,2,3,6،2،6،70-تتراهیدروپیریدین و همچنین نشان دادن افزایش mRNA و سطح پروتئین مارکرهای التهابی مانند COX-2 ، iNOS حساسیت بالایی دارند. ، IL-5 و TNF-؟ [2] علاوه بر این ، کبد های Nrf2؟ /؟ موشهایی که با رژیم کمبود متیونین و کولین روبرو شده اند بیان mRNA Cox71 و iNOS fold 2 برابر بیشتر از موشهای WT با همان رژیم دارند ، که نقش ضد التهابی Nrf2 را نشان می دهد [2]. اخیراً ، کیم و همکاران نشان داد که پیروات اتیل فیتوشیمیایی با کاهش بیان iNOS از طریق سیگنالینگ Nrf65 در سلول های BV300 ، اثرات ضد التهابی و ضد اکسیداتیونی خود را اعمال می کند. آنها نشان دادند که اتیل پیروات باعث جابجایی هسته ای Nrf72 می شود ، که در نهایت از تعامل بین p2 و p2 جلوگیری می کند و منجر به کاهش بیان iNOS می شود [2]. علاوه بر این ، آنالوگ کاربازول LCY-73-CHO NrfXNUMX را فعال کرده و باعث جابجایی هسته ای آن می شود و منجر به سرکوب بیان COXXNUMX و iNOS [XNUMX] در سلولهای عضله صاف عروقی آئورت موش می شود.
نقش متناقض Nrf2 در تنظیم فعالیت NLRP3 iIflammasome
خانواده NLR ، دامنه پیرین حاوی 3 (NLRP3) التهاب یک مجموعه چند پروتئینی است که به عنوان یک گیرنده تشخیص پاتوژن (PRR) عمل می کند و طیف گسترده ای از سیگنالهای استرس اکسیداتیو میکروبی مانند الگوهای مولکولی مرتبط با پاتوژن (PAMPs) را تشخیص می دهد ، مولکول های الگوی مولکولی مرتبط (DAMP) و ROS [74]. التهابات فعال NLRP3 باعث تجزیه کاسپاز -1 و ترشح سیتوکین اینترلوکین -1 پیش التهابی می شود؟ (IL-1؟) که در نهایت روند مرگ سلولی معروف به پیروپتوز را ایجاد می کند و از میزبان در برابر طیف وسیعی از عوامل بیماریزا محافظت می کند [75]. با این حال ، فعال شدن نابجای التهاب همراه با بیماری های غلط پروتئینی مانند انسفالوپاتی های اسفنجی شکل قابل انتقال ، بیماری آلزایمر ، بیماری پارکینسون و همچنین دیابت نوع 2 [76] ، سرطان [77] ، نقرس و تصلب شرایین [78] است.
مشاهدات اخیر گروه Rong Hu در رابطه با Nrf2 با تنظیم منفی التهاب نشان داد که ، Nrf2 باعث بیان NQO1 می شود که منجر به مهار فعال شدن التهاب NLRP3 ، تجزیه کاسپاز 1 و IL-1 می شود؟ تولید در ماکروفاژها. علاوه بر این ، یک فعال کننده Nrf2 شناخته شده ، ترت بوتیل هیدروکینون (tBHQ) با فعال کردن ARE به روش وابسته به Nrf3 ، رونویسی NLRP2 را منفی تنظیم می کند [79]. علاوه بر مشاهدات فوق ، همان گروه همچنین نشان داده شده است که ، دی متیل فومارات (DMF) از طریق فعال کردن مسیر سیگنالینگ Nrf2 که در انتقال هسته Nrf2 و مهار مونتاژ التهاب NLRP3 درگیر است ، از کولیت ناشی از DSS جلوگیری می کند [80].
یک سری آزمایشات با استفاده از ترکیبات طبیعی و مصنوعی نیز اثر مهاری Nrf2 را در فعال سازی التهاب NLRP3 نشان داده است. به عنوان مثال ، درمان اپی گالوكاتچین-3-گالات (EGCG) در موش كلیه نفریت نشان داده است كه باعث كاهش فعال سازی التهاب NLRP3 در كلیه می شود كه با واسطه مسیر سیگنالینگ Nrf2 انجام می شود [81]. به همین ترتیب ، سیترال (3,7،2,6-دی متیل-3،2-اکتادینال) ، یک ترکیب اصلی فعال در یک داروی گیاهی چینی Litsea cubeba ، از طریق سیگنالینگ آنتی اکسیدانی Nrf82 در مدل موش های سریع و شدید التهاب لوپوس (ASLN) باعث فعال شدن التهاب NLRP2 می شود [3] به طور مشابه ، بیوکانین با فعال کردن مسیر Nrf83 و مهار فعال سازی التهاب NLRP2 در موش های نر BALB / c در برابر آسیب کبدی ناشی از LPS / GalN محافظت می کند [1]. علاوه بر این ، نشان داده شد که منگیفرین میزان تنظیم Nrf3 و HO-1 را به روشی وابسته به دوز تنظیم کرده و NLRP1 ، ASC ، کاسپاز -84 ، IL-XNUMX کبدی ناشی از LPS / D-GalN را مهار می کند؟ و TNF-؟ بیان [XNUMX].
علی رغم تنظیم منفی NLRP3 توسط Nrf2 ، عملکرد التهابی NLRP3 و AIM2 را نیز فعال می کند. هایتائو ون و همکارانش کشف کردند ، Nrf2؟ ماکروفاژهای موش فعال سازی معیوب NLRP3 و AIM2 Inflammasome را نشان داده اند اما التهاب NLRC4 را نشان نمی دهند [85]. جالب توجه است ، این مشاهده عملکردهای ناشناخته Nrf2 را در زمینه بیماری های مرتبط با التهاب نشان می دهد. از این رو مطالعه بیشتر برای کشف مکانیزمی که Nrf2 عملکرد التهاب را قبل از در نظر گرفتن آن به عنوان یک هدف درمانی فعال می کند ، بسیار مهم است.
سرکوب رونویسی سیتوکین پروتئین التهابی توسط Nrf2
یک تحقیق بسیار اخیر مبتنی بر رسوب ایمنی کروماتین (ChIP) -seq و نتایج ChIP-qPCR در ماکروفاژهای موش نشان داد که Nrf2 به مناطق پروموتر سایتوکاین های پیش التهابی مانند IL-6 و IL-1 متصل می شود؟ و جذب RNA Pol II را مهار می کند. در نتیجه ، RNA Pol II قادر به پردازش فعال سازی رونویسی IL-6 و IL-1 نیست؟ که در نهایت منجر به مهار بیان ژن می شود. برای اولین بار ، گروه ماسایوکی یاماموتو مکانیزم جدیدی را کشف کرد که توسط آن Nrf2 نه تنها ژن های پایین دست خود را از طریق ARE غیر فعال می کند بلکه با جلوگیری از جذب RNA Pol II ، فعالیت رونویسی ژن های خاص را با یا بدون ARE سرکوب می کند [50].
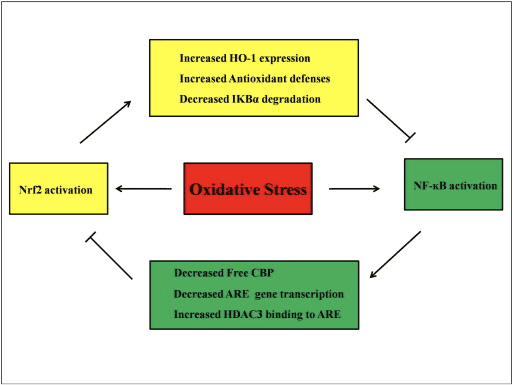
Crosstalk بین مسیرهای Nrf2 و NF-؟ B
NF-؟ B یک مجموعه پروتئینی است که مسئول رونویسی DNA است که تقریباً در همه انواع سلولهای حیوانی یافت می شود و در فرایندهای مختلفی مانند التهاب ، آپوپتوز ، پاسخ ایمنی ، رشد سلول و رشد نقش دارد. p65 ، یک پروتئین Rel از خانواده NF-؟ B ، دارای یک دامنه فعال سازی است در حالی که p50 ندارد و برای فعال سازی رونویسی به هترو دیمریزاسیون با پروتئین Rel نیاز دارد. در طی استرس اکسیداتیو ، I؟ B کیناز (IKK) فعال شده و باعث فسفوریلاسیون I؟ B می شود و در نتیجه منجر به آزاد سازی و انتقال هسته NF-؟ B می شود. NF-؟ B باعث رونویسی واسطه های پیش التهابی مانند IL-6 ، TNF- ؟، iNOS ، IL-1 و چسبندگی داخل سلولی COX-2 می شود.
تنظیم غیر طبیعی NF-؟ B به آرتریت روماتوئید ، آسم ، بیماری التهابی روده و گاستریت ناشی از عفونت هلیکوباکتر پیلوری مرتبط شده است [86]. در حال حاضر در نظر گرفته شده است که فعالیت NF-kB به طور عمده از سه جنبه روی مسیر سیگنالینگ Keapl / Nrf2 / ARE تأثیر می گذارد: اول ، Keap1 IKK را تخریب می کند؟ از طریق ubiquitination ، بنابراین فعالیت NF-؟ B را مهار می کند [87]. دوم ، روند التهابی باعث ایجاد واسطه های التهابی مانند COX2 مشتق شده از سیکلوپنتنون پروستاگلاندین 15d-PGJ2 می شود ، یک الکتروفیل قوی که با Keap1 واکنش نشان می دهد و Nrf2 را فعال می کند ، بنابراین رونویسی ژن با مهار همزمان فعالیت NF-kB آغاز می شود [58] ، [88] ( شکل 3 A ، B) سوم ، NF-؟ B می تواند با فعال کننده رونویسی رونوشت Nrf2 CBP [89] ، [90] ترکیب شود (شکل 3 C ، D).
شکل 3 عبور متناوب بین مسیرهای Nrf2 و NF-؟ B. (A) Keap1 IKK را به سمت Ubiquitination و تخریب پروتئازوم با واسطه CUL3 هدایت می کند که در نهایت منجر به مهار فسفوریلاسیون NF-؟ B می شود و این مکانیسم همچنین به عنوان اتصال رقابتی Nrf2 و IKK با Keap1 عمل می کند. (B) استرس اکسیداتیو IKK را فعال می کند که NF-؟ B را فسفریله می کند ، منجر به انتقال آن به هسته و فعال شدن سیتوکین های پیش التهابی مانند COX-2 می شود. محصول نهایی COX-2 معروف به 15d-PGJ2 به عنوان القا کننده Nrf2 عمل می کند که در نهایت منجر به سرکوب استرس اکسیداتیو می شود. (C) Nrf2 با کوفاکتور رونویسی CBP همراه با ماف کوچک و سایر ماشین آلات رونویسی پیوند می یابد تا بیان ژن ARE را آغاز کند. (D) هنگامی که NF-؟ B به روشی رقابتی با CBP متصل می شود ، اتصال CBP با Nrf2 را مهار می کند ، که منجر به مهار فعال سازی Nrf2 می شود.
فرض بر این است که مسیرهای سیگنالینگ Nrf2 و NF-؟ B برای کنترل رونویسی یا عملکرد پروتئین های هدف پایین دست در تعامل هستند. در توجیه این فرض مثالهای زیادی نشان می دهد که فعال سازی و مهار مستقیم یا غیر مستقیم بین اعضای مسیرهای Nrf2 و NF-؟ B رخ می دهد (شکل. 4). در پاسخ به LPS ، Nrf2 knockdown به طور قابل توجهی فعالیت رونویسی NF-؟ B و رونویسی ژن وابسته به NF-؟ B را افزایش می دهد ، نشان می دهد که Nrf2 مانع فعالیت NF-؟ B می شود [60] ، [91]. علاوه بر این ، افزایش بیان وابسته به Nrf2 در پایین دست HO-1 فعالیت NF-؟ B را مهار می کند. هنگامی که سلولهای سرطانی پروستات به طور خلاصه در معرض؟ -توکوفریل سوکسینات قرار بگیرند ، مشتق ویتامین E ، بیان HO-1 تنظیم مجدد می شود. محصولات نهایی HO-1 از انتقال هسته NF-؟ B جلوگیری می کنند [92]. این مطالعات in vivo نشان می دهد که Nrf2 منفی مسیر سیگنالینگ NF-kB را تنظیم می کند. LPS فعالیت اتصال DNA NF-؟ B را تحریک می کند و سطح زیر واحد p65 NF-؟ B در عصاره های هسته ای از ریه های Nrf2؟ /؟ به طور قابل توجهی بالاتر است. از موش WT ، نشان دهنده نقش منفی Nrf2 در فعال سازی NF-؟ B است. علاوه بر این ، Nrf2؟ /؟ فیبروبلاست های جنین موش تحت درمان با LPS و TNF-؟ فعال سازی برجسته تر NF-؟ B ناشی از فعال سازی IKK و I؟ B-؟ تخریب [60]. و ترشح ویروس سنسیسیال تنفسی به طور قابل توجهی کاهش می یابد در حالی که فعالیت اتصال DNA NF-؟ B در Nrf2؟ /؟ موشها با موشهای WT مقایسه شدند [93]. نفریت لوپوس ناشی از Pristane در Nrf2؟ /؟ موشهایی که تحت درمان با سولفورافان قرار گرفتند ، آسیب کلیوی شدید و تغییرات پاتولوژیک و همچنین افزایش بیان iNOS و فعال شدن NF-؟ B در مقایسه با WT دارند ، که نشان می دهد Nrf2 با مهار مسیر سیگنالینگ NF-؟ B و پاکسازی ROS باعث بهبود نفریت لوپوس می شود [94 ] فعالیت NF-؟ B همچنین هنگامی اتفاق می افتد که سلول ها با یک القا کننده Nrf2 همراه با LPS و TNF-؟ تحت درمان قرار گیرند. به عنوان مثال ، یک مشتق کالکون مصنوعی از طریق القای بیان HO-1 در سلولهای HT-29 اپیتلیال روده انسان ، فعال NF- β B ناشی از TNF -؟ را مهار می کند [62]. سرکوب انتقال NF-؟ B و فعالیت اتصال DNA و همچنین سرکوب بیان iNOS در سلولهای کبدی هنگامی که موشهای صحرایی F344 با 3H-1,2،3-dithiole-3-thione (D95T) تحت درمان قرار می گیرند یافت می شود [XNUMX]. پس از درمان همزمان با سولفورافان و LPS ، بیان ناشی از LPS iNOS ، COX-2 و TNF-؟ در ماکروفاژهای خام 264.7 ، تنظیم نشده است ، پیشنهاد می شود که سولفورافان از طریق مهار اتصال DNA NF-؟ B دارای فعالیت ضد التهابی است [96]. اگرچه چندین مطالعه تجربی برای توضیح ارتباط بین مسیرهای Nrf2 و NF-؟ B انجام شده است ، اما نتایج متناقضی همچنان وجود دارد. هر دو مقررات مثبت و منفی بین Nrf2 و NF-kB [97] گزارش شده است. به طور معمول، الکتروفیل های شیمیایی 3H-1,2-dithiole-3-tion، sulforaphane و Triterpenoid CDDO-Me با مهار NF-kB و ژن های پایین [2]، [98]، [99] فعال Nrf100 را فعال می کنند. در مقابل، چندین عامل یا شرایط مانند ROS، LPS، تنش برشی جریان، LDL اکسید شده و سیگار کشیدن نشان داده اند که فعالیت های Nrf2 و NF-kB [97] را افزایش می دهد. علاوه بر این ، مطالعات in vivo نشان داده است که فعالیت NF-kB در کبد جدا شده از Nrf2؟ /؟ کاهش می یابد. موش و فعالیت اتصال NF-؟ B در Nrf2؟ /؟ کمتر است. در موش های Nrf2 + / + [101]. با این حال ، سلولهای اندوتلیال آئورت انسانی تحت درمان با نانوذرات آدنو ویروسی Nrf2 بدون تأثیر بر فعالیت NF-؟ B ژنهای پایین دست NF-؟
شکل 4 حلقه تنظیم Nrf2 و NF-؟ B. مسیر Nrf2 با جلوگیری از تخریب I؟ B-؟ باعث فعال شدن NF-؟ B می شود. و افزایش بیان HO-1 و دفاع آنتی اکسیدانی که ROS و مواد شیمیایی سم زدایی را خنثی می کند. در نتیجه ، فعال سازی NF-؟ B مرتبط با ROS سرکوب می شود. به همین ترتیب ، رونویسی با واسطه NF-؟ B فعال شدن Nrf2 را با کاهش reduces کاهش می دهدهستندرونویسی ژن و پروتئین اتصال دهنده CREB رایگان با رقابت با Nrf2 برای CBP. علاوه بر این ، NF-؟ B باعث افزایش جذب هیستون دی استیلاز (HDAC3) به منطقه ARE می شود و از این رو از فعال سازی رونویسی Nrf2 جلوگیری می شود.
فعال سازی مسیر سیگنالینگ nrf2 نقش مهمی در بیان آنزیم ها و ژن هایی که در سم زدایی اکسیدان های واکنشی نقش دارند، با افزایش ظرفیت آنتی اکسیدانی سلول های بدن انسان است. در حالی که بسیاری از مطالعات تحقیقاتی در حال حاضر در دسترس هستند، مکانیزم های قانونی در فعال سازی Nrf2 به طور کامل درک نمی شوند. نقش احتمالی مسیر سیگنالینگ nrf2 در درمان التهاب نیز یافت شده است. دکتر الکس جیمنز DC، CCST Insight
نقش Nrf2 در بیماریهای التهابی
مطالعات in vivo نشان داده است که Nrf2 نقش مهمی در بیماری های التهابی دارد که بر سیستم های مختلف تأثیر می گذارد. این شامل گاستریت، کولیت، آرتروز، پنومونی، آسیب کبدی، بیماری قلبی عروقی، بیماری های نوروژنیک و آسیب مغزی است. در این مطالعات ، Nrf2؟ /؟ حیوانات علائم شدیدتر التهاب و آسیب بافتی را نسبت به حیوانات WT نشان دادند. بنابراین، اعتقاد بر این است که مسیر سیگنالینگ nrf2 اثر محافظتی در بیماری های التهابی دارد. نصب داخل تراشه از الاستاز پانکراس خوکی باعث بیماری مزمن انسدادی ریوی، به ویژه آمفیزم می شود. موش های کمبود Nrf2 بسیار حساس به آمفیزم هستند و بیان HO-1، PrxI و ژن antiprotease SLPI در ماکروفاژهای آلوئولار رخ می دهد. Nrf2 به عنوان یک تنظیم کننده کلیدی در سیستم دفاع متخصص ماکروفاژ در برابر آسیب های ریه دیده می شود [102]. موشهای کمبود Nrf2 با آمفیزم ناشی از قرار گرفتن در معرض دود سیگار برای ماه های 6 نشان دهنده افزایش التهاب برونکلو آلوئولی، افزایش بیان علامت های نشانگر استرس اکسیداتیو در آلوئول و افزایش آپوپتوز سلول های سپتوم آلوئولار است، که نشان می دهد که Nrf2 به دلیل افزایش بیان آنتی اکسیدان ژن ها [102]، [103]. با اختلال Nrf2، التهاب هوايي و آسم با استفاده از آلرژي با استفاده از مجتمع اوبوآبومين، التهاب هوايي، واکنش بالاي هوايي، هيپرپلازي سلول ها و سطوح بالاي Th2 در لارو bronchoalveolar و اسپلنوسيت ها، افزايش مي يابد، در حالي که Signalising راه نفوذي نفوپي، ، Hypersecretion موکوس، و بیش از حد واکنش های هوازی و همچنین ایجاد بسیاری از ژن های آنتی اکسیدانی که جلوگیری از آسم [2] است. تزریق کاراژین به حفره پلورال باعث التهاب پلاسمایی می شود و انباشت 15d-PGJ2 در سلولهای التهابی Nrf2 محدود به ماکروفاژهای پریتونیک ماوس می شود. در طول فاز اولیه التهاب، 15d-PGJ2 فعال Nrf2 و تنظیم روند التهابی را از طریق القاء HO-1 و PrxI تنظیم می کند. مطالعه همچنین نشان داد که COX-2 دارای اثر ضد التهابی در فاز اولیه با تولید 15d-PGJ2 [105] است. تجویز دهانی 1٪ سدیم سدیم سولفات سدیم برای هفته 1 موجب کولیت در ارتباط با تغییرات بافتی می شود که شامل کوتاه شدن کریپت ها و نفوذ سلول های التهابی در بافت کولون است. برای محافظت از یکپارچگی روده در کولیت، Nrf2 می تواند نقش مهمی را در تنظیم سیتوکین های پروتئین التهابی و ایجاد القاء آنزیم های فسفات II [51] بازی کند. در یک مدل موش Nrf2 حذفی از سپسیس ریوی ناشی از LPS ، فعالیت NF-؟ B تأثیر سیتوکین های التهابی مانند COX-2 ، IL-113 ، IL-6 و TNF را تنظیم می کند. که برای شروع و ترویج التهاب ضروری هستند [60]. Nrf2 با تنظیم این عوامل التهابی، آسیب های التهابی را کاهش می دهد. در این مدل التهاب حاد، تنظیم مقادیر آنزیم های آنتی اکسیدان، سیتوکین های پروتئین التهابی و واسطه ها با مسیر سیگنالینگ Nrf2 سبب کاهش آسیب التهابی در حیوانات WT می شود. جالب توجه است، این نیز در موش هایی که با نامحدودی Nrf2 قرار دارند گزارش شده است و علائم آن در مقایسه با موش های WT به طور قابل توجهی تشدید شده است.
تحقیقات در مورد داروهای ضد التهابی وابسته به Nrf2
به طور خلاصه، ما در مورد آزمایشاتی که نشان می دهد که مسیر سیگنال Nrf2 نقش مهمی در بسیاری از زمینه های التهاب بازی می کند، مورد بحث قرار گرفته است، بنابراین عوامل ضد التهابی وابسته Nrf2 برای درمان بیماری های التهابی مهم هستند.
گیاهان منابع فوق العاده غنی از ترکیبات است که عامل فاکتور رونویسی Nrf2 را فعال می کنند، که منجر به تنظیم مقررات ژن های سیتوپروتئین می شود. به تازگی، چندین مطالعه به منظور بررسی اثرات عوامل مختلف ضد التهابی، عمدتا از مایع گیاهی انجام شده است. به عنوان مثال، کورکومین ماده فعال زردچوبه است و همچنین در مقدار کمی در زنجبیل یافت می شود. ایزوتیوسیانات ها، به ویژه فنیللیسوتیوسیانات ها از بروکلی، کرفس و سایر سبزیجات هستند. و آنتوسیانین ها از انواع توت ها و انگور [124] می باشند. مطالعات نشان داده اند که تمام این عوامل نه تنها آنتی اکسیدان های خوب هستند، بلکه اثرات ضد التهابی قوی را از طریق القاء Nrf2 [125]، [126] دارند. بنابراین، توسعه فعال کننده های جدید ضد التهابی Nrf2 از عصاره گیاه علاقه زیادی به تحقیقات پزشکی دارد.
در سال های اخیر ، آزمایش های زیادی بر روی حیوانات انجام شده است تا عملکرد این ترکیبات را تأیید کند. Artesunate عمدتا برای مالاریای شدید ، مالاریای مغزی و بیماری های خود ایمنی روماتیسمی استفاده می شود. همچنین در آسیب ریه های سپتیک موثر است. Artesunate بیان Nrf2 و HO-1 را فعال می کند ، و دومی برای جلوگیری از التهاب ، سیتوکین ها و لکوسیت های پیش التهاب را به داخل بافت کاهش می دهد [127]. تصور می شود ایزوویتکسین ، که از پوست برنج Oryza sativa استخراج می شود ، دارای خواص ضد التهابی و آنتی اکسیدانی است. با فعال کردن مسیر Nrf2 / HO-1 و مهار MAPK و NF-؟ B [128] نقش محافظتی در برابر آسیب حاد ریه ناشی از LPS دارد. فیماسارتان ، یک مسدود کننده گیرنده آنژیوتانسین II که اخیراً محبوب شده و در سیستم رنین-آنژیوتانسین عمل می کند ، فشار خون را کاهش می دهد. استفاده از فیماسارتان برای درمان موشهایی که انسداد مجرای ادرار ناشی از جراحی دارند ، از طریق تنظیم مجدد Nrf2 و مسیر آنتی اکسیدان و مهار RAS و MAPK ، استرس اکسیداتیو ، التهاب و فیبروز را کاهش می دهد [129]. Sappanone به طور گسترده ای در جنوب شرقی آسیا توزیع می شود ، جایی که از آن به عنوان داروی ضد آنفلوانزا ، ضد حساسیت و محافظت از نور استفاده می شود. Nrf2 را فعال کرده و NF-؟ B را مهار می کند و بنابراین ممکن است در درمان بیماری های مربوط به Nrf2- و / یا NF-؟ B مفید باشد [130]. Bixin استخراج شده از دانه های Bixin orellana برای بیماری های عفونی و التهابی در مکزیک و آمریکای جنوبی استفاده می شود. این واسطه های التهابی ، نشت مویرگی آلوئولار و آسیب اکسیداتیو را به روشی وابسته به Nrf2 کاهش می دهد تا آسیب ریه ناشی از تهویه را کاهش دهد و مورفولوژی طبیعی ریه را بازگرداند [131]. سایر ترکیبات گیاهی مانند اپی گالوکاتچین گالات ، سولفورافان ، رسوراترول ، لیکوپن و عصاره چای سبز از طریق مسیر سیگنالینگ Nrf2 اثرات درمانی بر بیماری های التهابی دارند [132] ، [133] ، [134]. اخیراً گزارش شده است که یک ماده شیمیایی شیمیایی دیگر ، اریودیکتیل ، که در میوه مرکبات وجود دارد ، دارای اثرات ضد التهابی و آنتی اکسیدانی بر آسیب کلیه ناشی از سیس پلاتین و آسیب حاد ریه ناشی از سپسیس با تنظیم Nrf2 ، مهار NF-؟ B و مهار آن است. بیان سیتوکین ها در ماکروفاژها [135] ، [136]. با این حال ، مواد شیمیایی شیمیایی متعددی نویدبخش بزرگی برای پیشگیری و درمان بیماریهای مختلف انسانی است و برخی از آنها قبلاً وارد مرحله آزمایشات بالینی شده اند (جدول 2).
این ترکیبات گیاهی سیگنالینگ nrf2 را به طور عمده در مواد الکتروفیلنی فعال می کنند که باعث تغییر پسماندهای سیستین Keap1 می شود که منجر به پیوند آزاد Nrf2 هسته ای با ARE می شود و موجب فعال شدن رونویسی ژن متناظر می شود.
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
نتیجه گیری
در حال حاضر ، تحقیقات زیادی در مورد نقش مسیر سیگنالینگ Nrf2 / Keap1 / ARE در التهاب متمرکز شده است. در میان آنزیم هایی که توسط Nrf2 تنظیم مجدد می شوند ، HO-1 یکی از آنزیم های نماینده پاسخ استرس است. HO-1 دارای خواص ضد التهابی و آنتی اکسیدانی برجسته ای است. به طور کلی ، مسیر سیگنالینگ Nrf2 همچنین به طور منفی سیتوکین ها ، فاکتورهای آزاد کننده کموکین ، MMP ها و سایر واسطه های التهابی تولید COX-2 و iNOS را تنظیم می کند ، که به طور مستقیم یا غیر مستقیم مسیرهای مربوطه NF-kB و MAPK و سایر شبکه های کنترل کننده التهاب را تحت تأثیر قرار می دهند. پیشنهاد می شود که مسیرهای سیگنالینگ Nrf2 و NF-؟ B برای تنظیم رونویسی یا عملکرد پروتئین های هدف پایین دست در تعامل باشند. سرکوب یا غیرفعال سازی فعالیت رونویسی NF-؟ B با واسطه از طریق Nrf2 احتمالاً در فاز اولیه التهاب رخ می دهد ، زیرا NF-؟ B سنتز جدید آرایه ای از واسطه های پیش التهابی را تنظیم می کند. با این حال ، هنوز محدودیت هایی در تحقیقات وجود دارد از جمله اینکه آیا ارتباطی بین Nrf2 و سایر مسیرهای سیگنالینگ مانند JAK / STAT وجود دارد ، اهمیت فعال کننده های فعلی Nrf2 مشتق شده از منابع طبیعی گیاهی در التهاب و نحوه بهبود فعالیت بیولوژیکی و هدف قرار دادن این ترکیبات را افزایش می دهد. اینها به اعتبار آزمایشی بیشتری نیاز دارند.
علاوه بر این، مسیر سیگنال دهی Nrf2 می تواند بیش از 600 ژن [163] را تنظیم کند، که بیش از 200 ژن پروتئین های محافظ سلولی [164] را رمزگذاری می کنند که همچنین با التهاب، سرطان، بیماری های تخریب کننده عصبی و سایر بیماری های اصلی مرتبط هستند [165]. شواهد رو به رشد نشان می دهد که مسیر سیگنال دهی Nrf2 در بسیاری از سرطان ها غیرقابل تنظیم است و منجر به بیان ناهنجار باتری ژن وابسته به Nrf2 می شود. علاوه بر این، التهاب نقش مهمی در بیماری های مرتبط با استرس اکسیداتیو به ویژه در سرطان دارد. استفاده از چندین فعال کننده Nrf2 برای مقابله با التهاب ممکن است منجر به بیان نابجای ژن های پایین دستی Nrf2 شود که باعث ایجاد انکوژن و مقاومت در برابر شیمی درمانی و/یا رادیو درمانی می شود. بنابراین، فعالکنندههای بسیار اختصاصی Nrf2 ممکن است برای به حداقل رساندن اثرات پلیوتروپیک آن ایجاد شوند. چندین فعال کننده Nrf2 بهبود قابل توجهی در عملکردهای ضد التهابی در بیماری های مرتبط با استرس اکسیداتیو نشان داده اند. بهترین نمونه از فعال کننده Nrf2 مورد تایید FDA و به طور گسترده برای درمان بیماری های التهابی مانند ام اس (MS) دی متیل فومارات است. Tecfidera (نام ثبت شده دی متیل فومارات توسط Biogen) به طور موثر برای درمان اشکال عود کننده مولتیپل اسکلروزیس در تعداد زیادی از بیماران استفاده می شود [152]. با این حال، اثربخشی استفاده از فعالکنندههای Nrf2 برای درمان بیماریهای التهابی نیازمند اعتبارسنجی بیشتر برای جلوگیری از اثرات مضر Nrf2 است. بنابراین، توسعه درمانهایی برای فعالیت ضد التهابی با واسطه Nrf2 میتواند تأثیر بالینی قابلتوجهی داشته باشد. مطالعات در حال انجام مسیر سیگنال دهی Nrf2 در سراسر جهان به توسعه عوامل درمانی بسیار هدفمند برای کنترل علائم التهاب و پیشگیری و درمان سرطان و همچنین بیماری های عصبی و سایر بیماری های عمده اختصاص دارد.
در نتیجه، Nrf2 سطوح استرس اکسیداتیو را در بدن انسان حس می کند و در نهایت به ترویج تنظیم آنتی اکسیدان و سم زدایی آنزیم ها و ژن ها کمک می کند. از آنجا که التهاب مزمن ناشی از افزایش سطوح استرس اکسیداتیو با بیماری های نورودنژراتیک مرتبط است، Nrf2 می تواند یک نقش اساسی را بازی کند در درمان مسائل بهداشتی مانند بیماری آلزایمر ، و غیره. دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
زانو درد یک علامت شناخته شده است که می تواند به دلیل آسیب دیدگی های مختلف زانو و / یا شرایط ، از جمله رخ دهدآسیب های ورزشی. زانو یکی از پیچیده ترین مفاصل در بدن انسان است زیرا از تقاطع چهار استخوان ، چهار رباط ، تاندون های مختلف ، دو مینیسک و غضروف تشکیل شده است. طبق آكادمی پزشكان خانواده آمریكا ، شایع ترین دلایل درد زانو شامل زیر گرفتگی كشكك ، تاندینیت استخوان کشکک یا زانوی جهنده و بیماری اوسگود - شلاتر است. اگرچه زانو درد به احتمال زیاد در افراد بالای 60 سال بروز می کند ، اما درد زانو می تواند در کودکان و نوجوانان نیز رخ دهد. درد زانو را می توان با استفاده از روش های RICE در خانه درمان کرد ، با این حال ، آسیب های شدید زانو ممکن است نیاز به مراقبت فوری پزشکی ، از جمله مراقبت های کایروپراکتیک داشته باشد.
بیماری های نورودنراتیو، مانند بیماری آلزایمر و بیماری پارکینسون، میلیون ها نفر از افراد در سراسر جهان را تحت تاثیر قرار می دهد. گزینه های مختلف درمان برای درمان نشانه های بیماری های نورودنراتیو در دسترس هستند، اگرچه نتایج اغلب محدود است. تحقیقات تحقیقاتی نشان داده است که استرس اکسیداتیو ناشی از عوامل داخلی و خارجی می تواند باعث ایجاد بیماری های نوروژنیک شود. این فاکتور رونویسی، Nrf2، تعیین شده است که به عنوان یک مکانیسم دفاعی مهم در برابر استرس اکسیداتیو عمل کند. هدف مقاله زیر این است که اثرات آن را نشان دهد Nrf2 در بیماری های نوروژنیک.
مدولاسیون پروتئواستاز توسط NRF2 فاکتور رونویسی
بیماری های نوروژنیک ناشی از انباشته شدن ترکیبات پروتئینی خاص است، که نشان می دهد ارتباط صمیمانه بین مغز آسیب دیده و از دست دادن پروتئاز است. پروتئواستاز به تمامی فرآیندهایی که سلولهای فراوانی و کمرنگ شدن پروتئوم را به وسیله یک شبکه وسیع کنترل می کند که تنظیمات مسیرهای سیگنالینگ، بیان ژن ها و سیستم های تجزیه پروتئین را ادغام می کند. این بررسی تلاش می کند تا یافته های مربوطه در مورد مدولاسیون رونویسی پروتئاز با استفاده از فاکتور رونویسی NRF2 (عامل هسته ای (2 مشتق شده از اریتروتیست) مانند 2) خلاصه شود. NRF2 به عنوان سازنده اصلی پاسخ سلول های آنتی اکسیدان به طور کلاسیک در نظر گرفته شده است، اگر چه در حال حاضر به عنوان یک مولفه اصلی از دستگاه انتقال برای حفظ پروتئاز است. همانطور که ما در مورد بحث می کنیم، NRF2 می تواند به عنوان یک توپی طراحی شود که سیگنال های اضطراری حاصل از انباشت پروتئین نادرست را جمع آوری می کند تا یک پاسخ رونویسی هماهنگ و قابل انطباق ایجاد کند. این کار با عملکرد NRF2 مربوط به کنترل ژن هایی که در حفظ فیزیولوژی اندوپلاسمی رتیکولوم، پروتئازوما و اتوفایگی انجام می شود، به دست می آید.
فاکتور هسته ای (2 مشتق از اریتروتیست) مانند 2 (NRF2) یک پروتئین پایه-لوسین-زیپ است که امروزه به عنوان یک تنظیم کننده اصلی از هوموتازای سلولی در نظر گرفته شده است. این کنترل بیان پایه و القاء القاء بیش از ژن های 250 است که در یک مشترک تقویت کننده CIS عمل نامیده می شود عنصر پاسخ آنتی اکسیدان (ARE) [1]، [2]، [3]، [4]، [5]. این ژنها در واکنشهای سم زدایی فازهای I، II و III، متابولیسم گلوتاتیون و پروکسیروردوکین / تیرودوکسین، تولید NADPH از طریق مسیر پنتوز فسفات و آنزیم مالیک، اکسیداسیون اسید چرب، متابولیسم آهن و پروتئازوز [6] شرکت می کنند. با توجه به این عملکردهای سیتوپروتئینی وسیع، ممکن است که یک اثر فارماکولوژیک در NRF2 ممکن است اثر مجرمان اصلی بیماریهای مزمن را، از جمله استرس اکسیداتیو، التهابی و پروتئوتوکسیک را کاهش دهد. نقش NRF2 در مدولاسیون دفاع آنتی اکسیدانی و حل التهاب در مطالعات متعدد (در [7] مورد بررسی قرار گرفته است). در اینجا، ما بر نقش آن در پروتئازوزیس تمرکز می کنیم، یعنی کنترل هومیوستاتیک سنتز پروتئین، تاشو، قاچاق و تخریب. نمونه هایی در زمینه بیماری های نوروژنیک ارائه می شود.
از دست دادن تأثیرات پروتئوساستیس فعالیت NRF2 در بیماریهای نوروژنیک
یک علامت مشخصه عمومی از بیماری های تخریب عصبی ، وجود تجمع نابجا در برخی پروتئین ها است. بنابراین ، جمع شده های پروتئین نادرست آلفا-سینوکلئین (؟ -SYN) در بیماری پارکینسون (PD) ، پلاک های ب -امیلوئید (A؟) و گره های عصبی فیبر فسفریله TAU در بیماری آلزایمر (AD) ، هانتینگین (Htt) در بیماری هانتینگتون (HD) ، سوپراکسید دیسموتاز 1 (SOD1) و پروتئین اتصال دهنده TAR DNA 43 (TDP-43) در اسکلروز جانبی آمیوتروفیک (ALS) ، پروتئین prion (PrP) در انسفالوپاتی های اسفنجی شکل و غیره. پروتئین های پروتئینی می توانند در چندین اثر داشته باشند مسیرهای سلولی ، که به نوبه خود ممکن است بر سطح NRF2 و فعالیت آنها تأثیر بگذارد.
لایه های مختلف نظارت دقیق فعالیت NRF2
در شرایط فیزیولوژیکی، سلول های کم پروتئین NRF2 را به دلیل گردش سریع خود نشان می دهد. در پاسخ به محرک های مختلف، پروتئین NRF2 انباشته می شود، وارد هسته می شود و رونویسی ژن های حاوی ARE را افزایش می دهد. بنابراین، مدیریت پروتئین NRF2 یک نقطه اصلی است که باید سیگنال های ورودی مثبت و منفی را ادغام کند. همان طور که در ادامه خواهیم گفت، NRF2 توسط مکانیسم های همپوشانی متنوع برای فعال سازی پاسخ سریع و کارآمد فعال می شود اما از سوی دیگر NRF2 می تواند احتمالا در مرحله دوم مهار شود تا پاسخ آن را خاموش کند.
از نقطه نظر کلاسیک ، فعال شدن NRF2 به عنوان نتیجه پاسخ سلولی به ترکیبات اکسیدان یا الکتروفیل در نظر گرفته شده است. در این راستا ، آداپتور Ubiquitin E3 لیگاز Kelch مانند ECH مرتبط پروتئین 1 (KEAP1) نقش اساسی دارد. جزئیات مولکولی در بخش 4.1 بیشتر توضیح داده خواهد شد. به طور خلاصه ، KEAP1 به دلیل باقی مانده های حیاتی سیستئین که منجر به Ubiquitination NRF2 و تخریب پروتئازومی می شود ، به عنوان یک سنسور اکسایش اکسایش عمل می کند. علاوه بر این مدولاسیون کلاسیک ، NRF2 عمیقا با سیگنالینگ وقایع تنظیم می شود. در واقع ، کینازهای مختلف برای فسفوریلاسیون و تنظیم NRF2 نشان داده شده است. به عنوان مثال ، NRF2 را می توان با پروتئین کینازهای فعال شده با میتوژن (MAPK) فسفریله کرد ، اگرچه سهم آن در فعالیت NRF2 نامشخص است [8] ، [9] ، [10] ، [11]. PKA کیناز و همچنین برخی از ایزوآنهای PKC [12] ، CK2 [13] یا Fyn [14] فسفریلات NRF2 که پایداری آن را تغییر می دهند. کارهای قبلی گروه ما گزارش کرده اند که گلیکوژن سنتاز کینس -3؟ (GSK-3؟) با حذف هسته و تخریب پروتئازومی NRF2 را مهار می کند [15] ، [25] ، [26] ، [27] ، [28] ، [29] ، [30]. جزئیات مولکولی در بخش 4.1 مورد بحث قرار خواهد گرفت. علاوه بر این ، NRF2 به انواع دیگر مقررات ارسال می شود. به عنوان مثال ، استیلاسیون NRF2 توسط CBP / p300 فعالیت آن را افزایش می دهد [17] ، در حالی که توسط miR153 ، miR27a ، miR142-5p و miR144 [16] یا متیلاسیون جزایر سیتوزین-گوانین (CG) در پروموتر NRF2 مهار می شود. [18]
تاثیر متقابل پروتئین بر مکانیزم های تنظیم کننده NRF2
در این بخش ما تمرکز خواهیم کرد که چگونه تجمع پروتئین غلط شده می تواند فعالیت NRF2 را تحت تاثیر قرار دهد و بعضی از مسیرهای ذکر شده در بالا را به عنوان مثال های تصویری ارائه دهد. اولا ما باید در نظر داشته باشیم که انباشت پروتئین به شدت با آسیب اکسیداتیو ارتباط دارد. در واقع، تجمع و تجمع پروتئین غلط شده باعث تولید غیر طبیعی از گونه های اکسیژن واکنشی (ROS) از میتوکندری و دیگر منابع [19] می شود. همانطور که در بالا ذکر شد، ROS به سیستم های KEP1 حساس به حساسیت به بازده تبدیل می کند که منجر به انتشار، تثبیت و محلی سازی NRF2 هسته می شود.
در مورد پروتئین پاتی ها ، یک نمونه از وقایع سیگنالینگ تنظیم نشده که ممکن است NRF2 را تحت تأثیر قرار دهد ، با بیش فعالی GSK-3 ارائه می شود؟ در آگهی. GSK-3؟ ، همچنین به عنوان TAU کیناز شناخته می شود ، در فسفوریلاسیون این پروتئین مرتبط با میکروتوبول شرکت می کند ، و در نتیجه تجمع آن ، ایجاد گره های عصبی الیاف و قطع حمل و نقل آکسون (بررسی شده در [20]). از طرف دیگر ، GSK-3؟ همانطور که در بالا ذکر شد به طور چشمگیری سطح و فعالیت NRF2 را کاهش می دهد. آبشار آمیلوئید اگرچه به طور گسترده ای پذیرفته نیست ، پیشنهاد می کند که سمی A؟ الیگومرها GSK-3 را افزایش می دهند؟ فعالیت همراه با بیش فسفوریلاسیون TAU و مرگ نورون [21] ، [22]. مدل های مختلفی برای توضیح چگونگی A وجود دارد؟ GSK3 را ترجیح می دهد؟ فعالیت. به عنوان مثال ، A؟ به گیرنده انسولین متصل شده و مسیرهای سیگنالینگ PI3K و AKT را که برای حفظ GSK-3 بسیار مهم هستند ، مهار می کند؟ توسط فسفوریلاسیون در باقی مانده N9 ترمینال آن غیرفعال می شود [23]. از طرف دیگر ، A خارج سلولی؟ با گیرنده های Frizzled ارتباط برقرار می کند ، سیگنالینگ WNT را مسدود می کند [24] و دوباره منجر به آزاد شدن فعال GSK-3 می شود. خلاصه ، الف؟ تجمع منجر به بیش فعالی غیرطبیعی GSK-3 می شود ، بنابراین یک پاسخ مناسب NRF2 را مختل می کند.
همانطور که در بخش زیر بحث شده است، پروتئین های غلط شده منجر به فعال سازی PERK و MAPK ها می شود که به نوبه خود تنظیم NRF2 [31]، [8]، [9]، [10]، [11] می شود. علاوه بر این، فعالیت CBP / p300 غیر قابل تنظیم در چندین پروتئینوپاتی [32] گزارش شده است و کاهش جهانی متیلاسیون DNA در مغزهای AD نیز نشان داده شده است [33]، بنابراین زمینه را برای بررسی ارتباط این یافته ها در تنظیم NRF2 فراهم می کند.
ما و دیگران در موارد necropsies از بیماران PD و AD مشاهده شده است افزایش مقدار پروتئین NRF2 و برخی از اهداف آن مانند heme oxygenase 1 (HMOX1)، NADPH quinone oxidase 1 (NQO1)، p62 و غیره، هر دو توسط ایمونوبولت و توسط ایمونوهیستوشیمی [34]، [35]، [36]، [37]، [38]، [39] است. تنظیم مقررات NRF2 در این بیماری ها به عنوان یک تلاش ناموفق مغز بیمار برای بازیابی مقادیر هوموستاتیک تفسیر شده است. با این وجود، مطالعه دیگری نشان می دهد که NRF2 عمدتا در سیتوپلاسم نورون های هیپوکامپ آمپلی فایر قرار دارد و این نشان می دهد که فعالیت رونویسی NRF2 در مغز [40] کاهش می یابد. قابل تصور است که ناسازگاری این مشاهدات مربوط به تغییرات در عواملی است که NRF2 را در طول مراحل پیشرفت عصب زایی کنترل می کند.
سه سیستم عمده به پروتئواستاز کمک می کند، یعنی پاسخ پروتئین باز (UPR)، سیستم پروتئازوم ubiquitin (UPS) و autophagy. بعد، ما شواهدی ارائه می دهیم که NRF2 را به عنوان یک سیگنال اورژانسی متصل به هیدرولیک فعال می کند که توسط ترکیبات پروتئین با دستگاه مشتق شده پروتئین فعال شده است.
NRF2 مشارکت در پاسخ پروتئین Unfolded (UPR)
فعال سازی NRF2 در پاسخ به UPR
پروتئین اکسیداتیو تاشو در ER توسط تعدادی از مسیرهای مجزا هدایت می شود ، که بیشترین محافظت از آن شامل پروتئین دی سولفید-ایزومراز (PDI) و سولف هیدیل اکسیداز آندوپلاسمی اکسیدوراویدین 1 (ERO1؟ و ERO1؟ در پستانداران) به عنوان اهدا کننده دی سولفید است. به طور خلاصه ، PDI به دلیل کاهش و اکسیداسیون آمینو اسیدهای سیستئین خود ، تشکیل و شکستن پیوندهای دی سولفید بین باقی مانده های سیستئین درون پروتئین ها را کاتالیز می کند. PDI با عملکرد آنزیم خانه داری ERO1 بازیافت می شود که پیوندهای دی سولفید را مجدداً به PDI وارد می کند [41]. اکسیژن مولکولی پذیرنده الکترون پایانی ERO1 است که مقادیر استوکیومتری پراکسید هیدروژن را برای هر باند دی سولفید تولید می کند [42]. پراکسیدازها (PRX4) و گلوتاتیون پراکسیدازها (GPX7 و GPX8) آنزیم های کلیدی برای کاهش پراکسید هیدروژن در ER هستند. وقتی این سیستم اکسیدو احیا کننده به درستی کار نکند ، تجمع غیر طبیعی پروتئین های غلت خورده در ER رخ می دهد و مجموعه ای از سیگنال ها به نام پاسخ پروتئین باز نشده (UPR) به سیتوپلاسم و هسته منتقل می شود تا مجدداً هموستاز ER ایجاد شود. [43] سه پروتئین مرتبط با غشا برای سنجش استرس ER در یوکاریوت ها مشخص شده است: فاکتور رونویسی فعال 6 (ATF6) ، ER eIF2 پانکراس؟ کیناز (PERK ، همچنین پروتئین کیناز شبیه ER کیناز فعال شده با RNA دو رشته) و کیناز 1 نیاز به اینوزیتول (IRE1). دامنه مجرای هر سنسور به یک پروتئین تنظیم شده گلوکز (GRP78 / BIP) به نام چاپرون 78 کیلو دالتون متصل است. BIP با متصل شدن به استرس ER برای اتصال پروتئینهای شکسته نشده ، منجر به فعال شدن سه سنسور می شود [44].
NRF2 و NRF1 همولوگ آن، همچنین مربوط به پاسخ آنتی اکسیدانی هستند، در انتقال انتقال UPR به هسته شرکت می کنند. در مورد NRF1، این پروتئین در غشای ER واقع شده است و در حال انتقال اتم های هسته ای به علت deglycosylation یا شکاف است. سپس، فعال سازی UPR منجر به پردازش NRF1 و تجمع هسته ی حاصل شده در بخش هسته می شود. با این حال، توانایی transactivating ژن های حاوی ARE از این قطعه NRF1 هنوز در حال بحث [45] است.
گلوور کاتر و همکارانش فعال سازی ارتوپدی NRF2 از C. elegans، SKN-1، با عوامل استرسزا متفاوت ER. افزایش بیان SKN-1 بستگی به واسطه های مختلف UPR، از جمله IRE1 یا PERK کرم ها [46] بود. در سلولهای کمبود PERK، اختلال سنتز پروتئین منجر به انباشت پراکسید های اندوژن و آپوپتوز بعد [47] می شود. اثر بخشی PERK برای محافظت از ER از این پراکسیدها ممکن است NRF2 باشد، زیرا گزارش شده است که PERK فسفوریلات NRF2 را در Ser40 می کند و از تخریب آن توسط KEAP1 [31] جلوگیری می کند. القاء ASK1 نیز احتمالا در این مسیر نقش مهمی را در اثر فعالیت کیناز IRA2 [1] ایفا می کند. اگر چه نقش MAPK ها در تنظیم NRF48 هنوز بحث برانگیز است، اخیرا پیشنهاد شد که مسیر IRE2-TRAF1-ASK2-JNK ممکن است NRF1 [2] (شکل 49) را فعال کند. جالب توجه است که در سلول های C. elegans و سلول های انسانی، شواهد جدید نشان می دهد که سولفنیلات سستیون IRE1 کیناز در حلقه فعال شدن آن، مانع از UPR ارزیابی IRE1 می شود و واکنش آنتی اکسیدانی p1 را به وسیله NRF38 آغاز می کند. داده ها حاکی از آن است که IRE2 یک عمل باستانی به عنوان یک نگهبان سیتوپلاسمیک دارد که p1 و NRF38 [2] را فعال می کند.
شکل 1 تنظیم NRF2 توسط UPR. انباشت پروتئین های باز و یا غلط شده داخل رتینوپلاستی اندوپلاسمی می تواند پاسخ پروتئینی باز شده (UPR) را آغاز کند. اولا، BIP chaperone از دامنه intraluminal از سنسورهای ER IRE1 و PERK آزاد می شود تا پروتئین های پیچیده / ناسازگار را درگیر کند. این باعث می شود دیمرزاسيون و ترانسفورفوراسيون آنها را در دامنه های سيتوکسول انجام دهد. فعال سازی PERK منجر به فسفوریلاسیون NRF2 مستقیم در Ser40، منجر به انتقال NRF2 به هسته و فعال سازی ژن هدف می شود. فعال سازی IRE1 باعث جذب TRAF2 و پس از آن فسفوریلاسیون و فعال سازی ASK1 و JNK می شود. همانطور که JNK گزارش شده است که فسفوریلیت می کند و NRF2 را فعال می کند، منطقی است که فعال شدن IRE1 باعث افزایش فعالیت NRF2 شود.
بسیاری از مطالعات در مورد القای UPR با مهارکننده پروتئین گلیکوزیلاسیون تونیکامایسین انجام شده است. به نظر می رسد NRF2 برای جلوگیری از مرگ سلولهای آپوپتوتیک ناشی از تونیکامایسین ضروری است [31] و فعال شدن آن در این شرایط توسط تخریب اتوفاژی KEAP1 انجام می شود [51]. بر این اساس ، خاموش کردن بیان NRF2 در shRNA با واسطه در سلولهای TC-6 ، یک سلول سلول انسولینوما موش ، به طور قابل توجهی سمیت سلولی ناشی از تونیکامایسین را افزایش داد و منجر به افزایش بیان نشانگر استرس ER pro-apoptotic CHOP10 شد. از طرف دیگر ، فعال سازی NRF2 توسط 1,2،3-dithiole-3-thione (D10T) سمیت سلولی تونیکامایسین را کاهش داده و بیان CHOP52 و PERK را ضعیف می کند [2]. جالب توجه است ، نورونهای بویایی ارائه شده به کاربرد سیستمیک تونیکامایسین NRF1 را به موازات سایر اعضای UPR مانند CHOP ، BIP ، XBP53 افزایش می دهند [2]. این نتایج به مطالعات in vivo گسترش یافته است ، زیرا تزریق بطنی جانبی تونیکامایسین در موش صحرایی باعث بیان PERK و NRF42 در هیپوکامپ همراه با نقص شناختی قابل توجه ، افزایش فسفوریلاسیون TAU و رسوبات A؟ 54 می شود [XNUMX].
NRF2 تنظیم ژن های کلید برای حفظ فیزیولوژی اورژانس را تنظیم می کند
لومن ER به منظور حفظ شیمی دی سولفید به منبع فراوان GSH از سیتوزول نیاز دارد. NRF2 آنزیم های مهم متابولیسم GSH در مغز را تعدیل می کند ، مانند حمل سیستین / گلوتامات ، α-گلوتامات سیستئین سنتاز (؟ -GS) ، زیر واحد های کاتالیزوری و تعدیل کننده گلوتامات-سیستئین لیگاز (GCLC و GCLM) ، گلوتاتیون ردوکتاز (GR) و گلوتاتیون پراکسیداز (GPX) (بررسی شده در [55]). ارتباط NRF2 در نگهداری GSH در ER با این یافته تأیید می شود که فعال سازی دارویی یا ژنتیکی NRF2 منجر به افزایش سنتز GSH از طریق GCLC / GCLM می شود ، در حالی که با مهار بیان این آنزیم ها توسط NRF2-knockdown باعث تجمع آسیب دیده می شود پروتئین های موجود در ER منجر به فعال شدن UPR می شود [56].
در C. elegans چندین م ofلفه ژن هدف UPR که توسط SKN-1 تنظیم می شوند ، از جمله Ire1 ، Xbp1 و Atf6. اگرچه NRF2 بیان چندین ژن پراكسیداز (PRX) و گلوتاتیون پراكسیداز (GPX) را در پستانداران تنظیم می كند (فقط در [57] بررسی می شود) ، فقط GPX8 آنزیمی با موضع ER محلی است كه سیگنال بازیابی KDEL را در خود جای داده است. از دست دادن GPX58 باعث فعال شدن UPR ، نشت پراکسید هیدروژن مشتق شده از ERO8 به سیتوزول و مرگ سلول می شود. پراکسید هیدروژن مشتق شده از ERO1؟ فعالیت به دلیل عملکرد هماهنگ GPX1 و PRX8 نمی تواند از ER به سیتوزول پخش شود [4]. در این راستا ، تجزیه و تحلیل آرایه بیان ژن مسیر دفاعی آنتی اکسیدان با استفاده از RNA از نوع وحشی و بافت موش های پوستی NRF59 ، نشان داد که بیان GPX2 در غیاب NRF8 تحت تنظیم قرار گرفته است [2]. در همین راستا ، تجزیه و تحلیل نمونه برداری از نمونه های بیمار که از نئوپلاسم های میلوپرولیفراتیو ، پلی سیتمی یا میلوفیبروز رنج می برند ، بیماری هایی نیز با استرس اکسیداتیو و التهاب مزمن درجه پایین همراه هستند ، سطح بیان پایین تری از NRF60 و GPX2 را در مقایسه با افراد کنترل نشان می دهد [8]. هنوز مطالعاتی وجود ندارد که GPX61 به طور خاص در محافظت از مغز انسان دخیل باشد اما تجزیه و تحلیل رونوشت در موش نشان دهنده افزایش جبرانی GPX8 در پاسخ به سم پارکینسون MPTP است [8].
تاثیر NRF2 در تنظیم اختلال UPR در بیماری های نوروژنیک
بدرفتاری با آنزیم های PDI و فعال شدن مزمن UPR ممکن است به دنبال ایجاد یا تسریع رشد عصبی باشد. نورون های تحت تأثیر بیماری، مدل های حیوانی بیماری های نورودنژراتیو و همچنین بافت های انسانی پس از مرگ نشان دهنده تعدیل چندین نشانگر UPR در بسیاری از این اختلالات است. تغییر مسیر PDI / UPR در بیماری های نورودنژنتیک به خوبی در [63] مورد بررسی قرار گرفت، اما موارد زیر از نمونه های مغز پس از مرگ باید مورد توجه قرار گیرد. سطوح PDI در نورون های غلط گیر و در بدن Lewy از AD و PD به ترتیب [64]، [65] افزایش می یابد. PDI و ERP57 در CSF از بیماران ALS و در مغز از موارد CJD [66]، [67]، [68] تنظیم می شوند. BIP، PERK، IRE1 و ATF6 در نمونه هایی از بیماران مبتلا به AD، PD یا ALS [69]، [70]، [71]، [67] افزایش می یابد. BIP، CHOP و XBP1 در نمونه های مغز پس از مرگ از HD [72]، [73] افزایش می یابد. علاوه بر این، تنظیم مقادیر ERP57، GRP94 و BIP در بافتهای قشر از بیماران CJD [74] یافت شد. در مجموع، این شواهد نشان می دهد که انباشت پروتئین های غلط شده در parenchyma مغز منجر به فعال شدن مزمن و مزمن UPR می شود. جالب توجه است، یک مطالعه اخیر نشان می دهد که فعال شدن NRF2 توسط PERK در اوایل سال AD وجود دارد. در این مطالعه، نویسندگان، تجزیه و تحلیل کردند که آیا تغییرات میانجی کننده اکسیداتیو در NRF2 و UPR ممکن است باعث ایجاد وقایع اولیه در پاتوژنز AD با استفاده از سلول های خون محیطی انسان و یک مدل موس موش نژاد AD در مراحل مختلف بیماری باشد. افزایش استرس اکسیداتیو و افزایش pSer40-NRF2 در سلولهای تک سلولی خون محیطی انسان جدا شده از افراد مبتلا به اختلال شناختی خفیف مشاهده شد. علاوه بر این، گزارش شده اند که اختلالات هوموستاز کلسیم ER و نشانگرهای ER-stress در این سلول ها از افراد مبتلا به اختلال شناختی خفیف و AD [75] خفیف گزارش شده است.
تنظیم متقابل NRF2 و سیستم پروتئازوم یوبی کویتین (UPS)
یو پی اس سطح پروتئین NRF2 را تعدیل می کند
UPS در تخریب پروتئین های آسیب دیده یا ناسازگار شرکت می کند و سطوح مولکول های اصلی نظارتی را در سیتوزول و هسته کنترل می کند. هسته مرکزی این سیستم آنزیم multisubunit بزرگ است که شامل یک پروتئولیتیک فعال فعال به نام 20S است. پروتئازوم هسته 20S پروتئازوم را تجزیه می کند، اما اتصال به پروتئین های پروتئینی مختلف تنظیم کننده خاصیت و فعالیت بستر آن را تغییر می دهد. به عنوان مثال، اضافه کردن یک یا دو زیرمجموعه قانونی 19S به هسته 20S، پروتئازوم 26S را ایجاد می کند و خاصیت آن را نسبت به پروتئینهای 76 [77] بومی خود تغییر می دهد. تخريب پروتئازومي نياز به اتصال كووالنتال ubiquitin دارد. ترکیب ubiquitin از طریق یک مکانیزم آبشار سه مرحله ای حاصل می شود. اول، آنزیم فعال ubiquitin E1 ubiquitin را در واکنش مورد نیاز ATP فعال می کند. سپس، یک آنزیم E2 (پروتئین حامل ubiquitin یا آنوبیس کنجوابي ubiquitin) ubiquitin فعال از E1 را به سوبسترا منتقل می کند که به طور خاص به یک عضو از خانواده لیوزا پروتئین ubiquitin منتقل می شود، به نام E3. اگر چه سرنوشت دقیق پروتئین ubiquitinated به طبیعت زنجیره ubiquitin وابسته است، این روند به طور کلی منجر به تخریب پروتئازوم 26S [78] می شود.
E3-ligase KEAP1 یکی از بهترین بازدارنده های NRF2 شناخته شده است. مکانیزم تنظیمات KEAP1 ظرافتی را توضیح می دهد که چگونه سطوح NRF2 با نوسانات اکسیدان تنظیم می شود. در شرایط پایه NRF2 به تازگی سنتز شده توسط KEAP1 homodimer، که یک مولکول NRF2 در دو توالی آمینو اسید با کم (آسپارتیت، لوسین، گلیسین، DLG) و بالا (گلوتامات، ترئونین، گلوتامین، گلوتامات، ETGE) وابسته است. تعامل با KEAP1 کمک می کند تا NRF2 را به پروتئین CULLIN3 / RBX1 بسپارید، که منجر به ubiquitination آن و تخریب پروتئازوما بعد می شود. با این حال، اصلاح redox از KEAP1 مانع ارائه NRF2 به UPS نماینده CULLIN3 / RBX1 است. در نتیجه، NRF2 به تازگی Synthetized فرار از تخریب وابسته به KEAP1، در هسته تجمع می یابد و فعال ژن های حاوی ARE [79]، [80]، [81]، [82].
آداپتور E3-ligase؟ -TrCP نیز یک هومدایمر است که در رویدادهای سیگنالینگ مربوط به فسفوریلاسیون NRF2 توسط GSK-3؟ شرکت می کند. این کیناز فسفوریله باقیمانده های سرینی خاص NRF2 (آسپارتات ، سرین ، گلیسین ، ایزولوسین سرین ؛ DSGIS) را ایجاد می کند تا یک دامنه تخریب ایجاد کند که سپس توسط؟ -TrCP شناخته شده و برای تخریب پروتئازوم توسط یک مجموعه CULLIN1 / RBX1 برچسب گذاری شود. شناسایی اسیدهای آمینه خاصی که توسط GSK-3 فسفریله می شوند؟ در این دگرون با ترکیبی از جهش زایی سایت هدایت شده حوزه Neh6 ، الکتروفورز ژل 2D [15] ، [26] و طیف سنجی جرمی [83] انجام شد. در نتیجه ، مهار GSK-3؟ توسط داروهای بسیار انتخابی یا siRNA در برابر ایزوفرم GSK-3 منجر به افزایش سطح پروتئین NRF2 می شود. نتایج مشابهی با siRNA ها در برابر ایزوفرمهای 1-2؟ -TrCP پیدا شد. ثبات NRF2 به دنبال GSK-3؟ مهار در فیبروبلاست های جنینی موش کمبود KEAP1 و در جهش حذف NRF2 نابجا بیان شده و فاقد باقی مانده های حیاتی ETGE برای اتصال با میل بالا به KEAP1 ، بیشتر یک تنظیم مستقل از KEAP1 را نشان می دهد.
در زمینه بیماری های نورودژنراتیو ، ما می توانیم تعدیل NRF2 توسط UPS را به دو روش مختلف تصور کنیم. از یک طرف ، سیستم KEAP1 عدم تعادل ردوکس حاصل از تجمع پروتئین بهم ریخته را حس می کند ، در حالی که محور GSK-3 /؟ - TrCP به عنوان یک شرکت کننده فعال در انتقال سیگنالینگ تغییر می کند که با از دست دادن پروتئوز ، تغییر می کند (شکل 2).
شکل 2 UPS به شدت سطح NRF2 را کنترل می کند. تحت شرایط هوموستاتیک ، سطح NRF2 پایین با عملکرد آداپتورهای E3 لیگاز KEAP1 و؟ -TrCP حفظ می شود. از سمت چپ ، NRF2 از طریق نقوش ترکیبی کم (DLG) و بالا (ETGE) به دامنه های کلچ یک homodimer KEAP1 متصل می شود. از طریق دامنه BTB خود ، KEAP1 به طور همزمان به یک مجموعه CULLIN3 / RBX1 متصل می شود ، امکان استفاده مجدد و تخریب NRF2 توسط پروتئازوم 26 S را فراهم می کند. علاوه بر این ، GSK-3؟ فسفوریله های باقی مانده Ser335 و Ser338 از NRF2 برای ایجاد یک دامنه تخریب (DpSGIpSL) که سپس توسط آداپتور ubiquitin ligase؟ -TrCP شناخته می شود و برای تخریب پروتئازوم توسط یک مجموعه CULLIN3 / RBX1 برچسب گذاری می شود. درست است ، با قرار گرفتن در معرض گونه های اکسیژن واکنش پذیر یا الکتروفیل ها ، باقی مانده های حیاتی Cys در KEAP1 اصلاح می شوند ، بنابراین KEAP1 قادر به تعامل موثر با NRF2 یا CULLIN3 / RBX1 نیست و سپس این فاکتور رونویسی باعث افزایش نیمه عمر و فعالیت رونویسی آن نسبت به ژن های ARE می شود. مسیرهای سیگنالینگ که منجر به مهار GSK-3 می شوند ، از جمله فسفوریلاسیون AKT در Ser9 ، منجر به اختلال تخریب NRF2 توسط پروتئازوم ، تجمع و القای ژن های هدف می شود.
NRF2 فعالیت UPS را از طریق کنترل ترانسپورتی زیرمجموعه پروتئازوم افزایش می دهد
NRF2 بیان چندین زیر واحد پروتئازوم را تنظیم می کند ، بنابراین از سلول در برابر تجمع پروتئین های سمی محافظت می کند. بنابر تجزیه و تحلیل گسترده ریزآرایه از RNA کبد که با D2T القا کننده NRF2 تنظیم شده است ، به نظر می رسد بیست ژن مربوط به پروتئازوم و ubiquitination توسط NRF3 تنظیم می شوند. در یک مطالعه خلفی ، همان نویسندگان ثابت کردند که بیان اکثر زیرواحدهای پروتئازوم 84S در موشهای تحت درمان با D26T در کبد تا سه برابر افزایش یافته است. سطح پروتئین زیر واحد و فعالیت پروتئازوم به طور هماهنگ افزایش یافته است. با این حال ، در موشهایی که فاکتور رونویسی NRF3 مختل شده است ، هیچ القایی مشاهده نشده است. فعالیت پروموتر زیر واحد پروتئازوم PSMB2 (5S) با بیان بیش از حد NRF20 یا درمان با فعال کننده ها در فیبروبلاست های جنینی موش افزایش یافته و ARE در پروموزر پروگزیمال PSMB2 مشخص شد [5]. فعال سازی دارویی NRF85 منجر به افزایش سطح بیان زیر واحد پروتئازوم نمایندگی (PSMA2 ، PSMA3 ، PSMB6 و PSMB1) فقط در فیبروبلاست های انسانی غیر پیر پیر که حاوی NRF5 عملکردی هستند [2]. فعال سازی NRF86 در حین سازگاری با استرس اکسیداتیو منجر به بیان زیاد PSMB2 (1S) و PA20 می شود؟ زیر واحد (یا S28 ، تنظیم کننده پروتئازوم) [11]. علاوه بر این ، نتایج حاصل از سلول های بنیادی جنینی انسان نشان داد که NRF87 بیان پروتئین بلوغ پروتئازوم (POMP) ، یک چاپرون پروتئازوم را کنترل می کند ، که به نوبه خود تکثیر سلولهای بنیادی جنینی انسان را که خود تجدید می شوند ، سه تمایز لایه جوانه و برنامه ریزی مجدد سلول را تعدیل می کند [ 2] در مجموع ، این مطالعات نشان می دهد که NRF88 بیان اجزای اصلی UPS را تنظیم می کند و بنابراین فعالانه به پاکسازی پروتئین هایی کمک می کند که در غیر این صورت سمی هستند.
محور NRF2-UPS در بیماریهای نروژنیک
نقش یو پی اس در بیماری های نورودنراتیو، زمینه ای برای بحث های شدید است. مطالعات اولیه نشان داد که فعالیت پروتئازوما در انسداد انسانی بیماران مبتلا به بیماری های نورودنراتیو کاهش یافته است. با این حال، مطالعات دیگری که با استفاده از روشهای in vitro و in vivo فعالیتهای پروتئازوم بدون تغییر یا حتی افزایش یافته را بررسی کردند (در [89] بررسی شده است). یک توضیح ممکن برای این اختلاف، این است که سطح اجزاء یو پی اس ممکن است در طول پیشرفت بیماری تغییر کند و در مناطق مختلف مغز، همانطور که برای اهداف NRF2 پیشنهاد شده است.
علی رغم این اختلاف، باید توجه داشت که تنظیم مقررات ژنهای پروتئازوما حاوی ARE باعث تقویت UPS توسط افزایش پروتئین های سمی در مغز خواهد شد. در واقع، تخلیه NRF1، همچنین تعدیل کننده پاسخ آنتی اکسیدانی در سلول های عصبی، منجر به اختلال در فعالیت پروتئازوم و تولید عصبی می شود. آزمایشات ایمونوفیزیک کروماتین و تجزیه و تحلیل رونویسی نشان داد که PSMB6 توسط NRF1 تنظیم می شود. علاوه بر این، تجزیه و تحلیل بیان ژن منجر به شناسایی NRF1 به عنوان یک رژیم اصلی رونویسی ژن پروتئازوم در نورون، نشان می دهد که اختلالات در NRF1 ممکن است به پاتوژنز بیماری های نورودژنراتیو [90] کمک کند. جالب توجه است که NRF1 و ایزوفرم طولانی آن به نام TCF11 نشان داده شده است که ژن پروتئازوم حاوی ARE را پس از مهار پروتئازوما در یک حلقه بازنگری برای جبران کاهش فعالیت پروتئولیتیک [91]، [92] تنظیم می کنند.
در ارتباط با NRF2، در کاهش میان سطوح NRF2، RPT6 (19 S) و PSMB5 (20 S) در میان میانی موشهای کمبود DJ-1 که با پاراکوت نوروکسین [93] درمان می شوند، ارتباط وجود دارد. علاوه بر این، ترکیب سولفورفان طبیعی (SFN) یک تصویر قوی تر از NRF2 را به عنوان یک مدولاتور شدید یو پی اس می دهد. آزمایش های درون آزمایش با neuroblastoma murine Neuro2A سلول نشان داد افزایش بیان subunits کاتالیزوری پروتئازوم، و همچنین فعالیت های پپتیداز آن در پاسخ به SFN. این دارو از سلول های هیدروژن پراکسید هیدروکسید سدیم و اکسیداسیون پروتئین به نحوی وابسته به عملکرد پروتئازوما [94] محافظت می کند. علاوه بر این، لیو و همکارانش یک موسس گزارشگر را برای نظارت بر فعالیت UPS در پاسخ به SFN در مغز استخدام کردند. این موش ها در همه جا پروتئین فلورسانس سبز (GFP) را به یک سیگنال تخریب سازنده تبدیل می کند که باعث تخریب سریع آن توسط یو پی اس (GFPu) می شود. در مغزهای مغزی، SFN سطح GFPu را با افزایش موازی در فعالیت های کیموتریپسین (PSMB5)، کپاز (PSMB2) و فعالیت های ترپسین (PSMB1) پروتئازوم 20 S کاهش می دهد. علاوه بر این، درمان سلول های حاصل از هانتینگتون با SFN نشان داد که فعال شدن NRF2 باعث افزایش تشدید mHtt و کاهش سمیت سلول های mHtt [95] می شود. مکانیسم اصلی عمل SFN از طریق القاء NRF2 [96] است. سهم خاص NRF2 باید با استفاده از سیستم NRF2-null در مطالعات بیشتری مورد توجه قرار گیرد.
ارتباط عملکردی بین NRF2 و Macroautophagy
سطح پروتئین NRF2 توسط پروتئین آداپتور P62 مدولاسیون می شود
Autophagy به تخریب اجزای سیتوزول داخل لیزوزوم اشاره دارد. این فرآیند برای پاکسازی پروتئین های طولانی مدت و ناسازگار و همچنین ارگان های آسیب دیده استفاده می شود. پیوند مستقیم بین NRF2 و autophagy در ارتباط با پروتکل آداپتور p62، همچنین به نام SQSTM1 [97]، [98]، [99]، [100]، [101]، مشاهده شد. این پروتئین پروتئین ubiquitinated را به دستگاه های تجزیه پروتئازوم و لیزوزومی مغلوب می کند و پروتئین های آسیب دیده را به ارقام قبل از تخریب آنها منتقل می کند. P62 یک دامنه ubiquitin مرتبط (UBA) را برای اتصال به پروتئین های ubiquitinated و یک منطقه متداول LC3 (LIR) برای ادغام با غشای اتوآوآگاوسوم از طریق گیرنده اتوفاژی LC3 ارائه می دهد.
اگرچه القا N NRF62 و ژنهای هدف آن با واسطه p2 اولین بار در سال 2007 گزارش شد [102] ، اما مکانیسم مولکولی تا زمان کشف اثر متقابل آن با KEAP1 کاملاً درک نشده بود [103] ، [98] ، [99] ، [100] ] ، [101]. کوماتسو و همکاران در منطقه P1 منطقه متقابل KEAP62 (KIR) را شناسایی کردند که KEAP1 را در همان جیب سطح اصلی NRF2 و با میل اتصال مشابه نقوش ETGE در NRF2 متصل می کند ، پیشنهاد رقابت بین p62 و NRF2. فسفوریلاسیون Ser351 در نقش KIR در p62 (349-DPSTGE-354) نشان داده شد که میل آن به KEAP1 را افزایش می دهد ، با اتصال NRF2 رقابت می کند و اجازه تجمع و رونویسی ژنهای هدف آن را می دهد [98] ، [99]. در واقع ، بیان بیش از حد p62 منجر به کاهش فراوانی در استفاده از NRF2 و تثبیت متعاقب آن و همچنین القای ژن های هدف آن شد [104]. برخی از کینازها برای شرکت در فسفوریلاسیون p62 پیشنهاد شده است. هدف پستانداران کمپلکس راپامایسین 1 (mTORC1) ممکن است نقش داشته باشد ، زیرا درمان با مهارکننده mTOR رپامایسین فسفوریلاسیون p62 و تنظیم پایین KEAP1 را تحت درمان آرسنیت سرکوب می کند. اخیراً نشان داده شده است كه كیناز 1 فعال شده با TGF (TAK1) همچنین می تواند p62 را فسفریله كند و باعث تخریب KEAP1 و تنظیم NRF2 شود. نویسندگان این مطالعه پیشنهاد می کنند که این یک روش برای تنظیم مجدد سلولی تحت شرایط پایدار است ، زیرا کمبود TAK1 باعث افزایش ROS در صورت عدم وجود هرگونه اکسید کننده برون زا در بافتهای مختلف موش به موازات کاهش سطح پروتئین NRF2 می شود [105 ]
یک ساختار p62 که فاقد دامنه UBA بود، هنوز قادر به اتصال KEAP1 بود، و این بدان معنی است که تعامل به KEAP1 ubiquitinated [101] بستگی ندارد. اما همولوگ p62 در Drosophila melanogaster، به نام Ref (2)، حاوی یک KIR موتیف نیست و به طور مستقیم با DmKEAP1 ارتباط برقرار نمی کند، اگر چه می تواند به DmKEAP1 ubiquitinated از طریق دامنه UBA متصل شود. علاوه بر این، DmKEAP1 می تواند به طور مستقیم با Atg8 (همولوگ با LC3 پستانداران) ارتباط برقرار کند. کمبود KEAP1 منجر به القاء Atg8 و autophagy وابسته به NRF2 orthologue CncC و مستقل در TFEB / MITF [106]. به نظر می رسد رابطه NRF2 و autophagy هر چند حفظ می شود، برجسته ارتباطات کاربردی آن است.
القاء NRF2 توسط p62 نتیجه هر دو رقابت برای اتصال KEAP1 و تضعیف KEAP1 در لیزوزوم است. خاموش کردن p62 با siRNA، دوام نیمه عمر KEAP1 به موازات کاهش در NRF2 و ژن هدف آن [101]. در توافق، تخفیف بیان p62 نشان دهنده افزایش سطح KEAP1 در مقایسه با موش های وحشی است. بسیار مرتبط، افزایش در سطوح KEAP1 تحت تاثیر مهارکننده های پروتئازام قرار نگرفت، اما تحت اتوفایگی ناشی از گرسنگی [107] کاهش یافت. در حقیقت، KEAP1 در سلولهای پستانداران در حوضچه های اتوفاییک تزئین شده با p62 و LC3 [99]، [100]، [103] وجود دارد. همه این داده ها نشان می دهد که KEAP1 جایگزین ماشین آلات ماکرو Avophagy است، اما این موضوع باید با توجه به وجود برخی از نتایج بحث برانگیز با جزئیات بیشتر تجزیه و تحلیل شود. سطح پروتئین KEAP1 در موش Atg7-null، یک عامل کلیدی از ماکرووفلوفیک [107] افزایش یافت، اما مهار داروی ماکئوفوفایگی با torin1، E64 / pepstatin یا بافیلومایسین موفق به تجمع KEAP1 [107]، [100] نشد. به طور کلی، این نتایج نشان می دهد که سطوح p62 افزایش یافته است و KEAP1 را به واکسن های اتوفاییک متصل می کند و احتمالا این باعث کاهش تجزیه Autophagic KEAP1 می شود که به NRF2 فعال می شود (شکل 3). دو مطالعه متفاوت گزارش دادند که SESTRINS اسید سولفنیک اسید نقش مهمی در این زمینه ایفا می کند. SESTRIN 2 با p62، KEAP1 و RBX1 تعامل دارد و تضعیف تخریب وابسته به p62 از فعال سازی KEAP1 و NRF2 از ژن های هدف [108]. مطالعه دیگری نشان داد که SESTRIN 2 با ULK1 و p62 ارتباط برقرار کرد و باعث تقویت فسفوریلاسیون p62 در Ser403 شد که باعث کاهش تخریب پروتئین های محموله از جمله KEAP1 [109] شد.
شکل 3 NRF2 سطح توسط پروتئین آداپتور p62 تنظیم می شود. فسفوریلاسیون Ser 351 در موتیف KIR از p62 (349-DPSTGE-354) توسط mTORC1، TAK1 یا سایر کینازها موجب افزایش وابستگی به اتصال به KEAP1 به علت شباهت به موتیف ETGE در NRF2 می شود. به عنوان یک نتیجه، p62 فسفرولیید جابجا کردن NRF2 و اتصال KEAP1. ماتریس LIR در p62 باعث ایجاد تعامل با LC3 در غشای اتوفوگاسوم می شود، به طوری که پیچیده p62-KEAP1 در نهایت در لسیوزوم تخریب می شود. به عنوان یک نتیجه NRF2 قادر به جمع آوری، انتقال به هسته و افزایش رونویسی از ژن های حاوی ARE، از جمله p62. این مکانیزم نظارتی پاسخ NRF2 قابل تحمل را فراهم می کند، زیرا KEAP1 باید به تازگی برای مهار فعالیت NRF2 سنتز شود.
مدولاسیون ژن های ماکرووفوفیگی توسط NRF2
NRF2 بیانات ژن های مربوط به ماکرو Avophagy را تنظیم می کند و همچنین برای UPR و UPS انجام می شود. نخستین شواهدی از مطالعاتی بود که نشان داد که بیان p62 در معرض الکتروفیل ها، ROS و اکسید نیتریک [110]، [111]، [112] ایجاد می شود. مکانیسم القاء چند سال بعد با یافته هایش که P62 حاوی ARE عملکردی در پروموتر ژن آن [99] است توصیف شد. در یک تحقیق اخیر، چندین ARE های کاربردی دیگر یافت شده و پس از تجزیه و تحلیل بیوانفورماتیک و آزمایش ChIP مورد تایید قرار گرفتند. علاوه بر این، فیبروبلاست های جنینی جنینی و نورون های کورتنی از موش هایی که از nf2 خارج می شوند، بیان کاهش بیان p62 را می دهند که می تواند با یک لنفوویروس بیانگر NRF2 نجات یابد. به طور مشابه، کمبود NRF2 باعث کاهش سطح p62 در نورون های آسیب دیده از هیپوکامپ موش [36]. بنابراين پيشنهاد شده است كه فعال شدن NRF2 باعث افزايش سطح p62 مي شود و منجر به تخريب KEAP1 مي شود و به تثبيت NRF2 بيشتر در يك حلقه بازخورد مثبت كمك مي كند. این مکانیزم غیر کانونی NRF2 القاء نیاز به تغییر در بیان ژن دارد و ممکن است پاسخ مناسب به استرس طولانی مدت سلولی باشد.
پروتئین NDP52 به رسمیت شناختن محموله توسط NRF2 تنظیم شده است. NDP52 به روش مشابه با p62 کار می کند، پروتئین ubiquitinated را تشخیص می دهد و با LC3 ارتباط برقرار می کند از طریق دامنه LIR، به طوری که در lysosomes تخریب می شود. پنج ARE های احتمالی در دنباله DNA پروموتر Ndp52 یافت شد. سه نفر از آنها با ساختارهای جهش یافته و آزمایشات ChIP به عنوان ضروری برای رونویسی Ndp2 [52] متصل به NRF113 شناسایی شدند. توجه داشته باشید که سطوح mRNA Ndp52 در هیپوکامپ موشهای نایفکس نیومکزیک کاهش می یابد. یکی از این توالی ها نیز در یک مطالعه مستقل به عنوان یک ARE [2] تنظیم NRF2 تایید شد.
با این حال، نقش NRF2 در مدولاسیون autophagy محدود به القاء از این دو پروتئین شناخته شدن محموله نیست. به منظور به دست آوردن بینش عمیق تر در نقش NRF2 در مدولاسیون ژن های مربوط به autophagy اضافی، گروه ما کروماتین پایگاه داده ایمن زدایی ENCODE برای دو پروتئین MAFK و BACH1، که به AREs تنظیم شده NRF2 متصل است. با استفاده از یک اسکریپت که از توالی ARE توافق JASPAR تولید شده است، ما چندین ARE های احتمالی را در بسیاری از ژنهای Autophagy شناسایی کردیم. دوازده از این توالی ها به عنوان NRF2 تنظیم شده اند AREs در نه ژن autophagy، بیان شده است که در فیبروبلاست های جنینی موش از موش های Knockout nof2 کاهش یافته است، اما می تواند توسط lentivirus بیان NRF2 بازسازی شود. مطالعه ما نشان داد که NRF2 بیان برخی از ژن های درگیر در مراحل مختلف فرآیند اتوفایی را فعال می کند، از جمله اتخاذ اتوفایگی (ULK1)، شناسایی محموله (p62 و NDP52)، تشکیل اتوفاهاوسوم (ATG4D، ATG7 و GABARAPL1)، انقطاع (ATG2B و ATG5 )، و پاکسازی autolysosome (ATG4D). در نتیجه، شار اتوفایگی در واکنش به پراکسید هیدروژن هنگامی که NRF2 غایب بود [36] اختلال نداشت.
معیارهای بیان ژنهای ماکروو اتوفافگی NRF2 در اختلالات نوروژنیک
نشان داده شده است که autophagy معیوب نقش مهمی در چندین بیماری شایع نانو [114] دارد و تخریب autophagy منجر به تولید عصبی در موش [115]، [116] می شود. موشهای نابودکننده Atg7 نشان داد که کمبود اتوفایگی منجر به انباشت p62 در بدنهای انسانی مثبت ubiquitin می شود. KEAP1 در این اجسام درون استخراج شد، منجر به تثبیت NRF2 و القاء ژنهای هدف [103] شد. مهم است که تجمع بیش از حد p62 همراه با پروتئین ubiquitinated در بیماری های نورودنژنتیک، از جمله AD، PD و ALS [117] شناسایی شده است. در حقیقت، نورونهایی که سطح بالایی از APP یا TAU بیماران AD را بیان می کنند، همچنین p62 و NRF2 هسته ای را بیان می کنند، که نشان می دهد تلاش آنها برای تخریب عناصر intraneuronal از طریق autophagy [36] است.
کمبود NRF2 تجمع پروتئین را در زمینه AD تشدید می کند. در حقیقت ، افزایش سطح TAU فسفریله و محلول در سارکوزیل در موشهای حذفی Nrf2 یافت می شود ، اگرچه در مقایسه با پس زمینه نوع وحشی هیچ تفاوتی در فعالیت های کیناز یا فسفاتاز قابل تشخیص نیست [113]. مهمتر از همه ، NDP52 به منظور محلی سازی با TAU در سلولهای عصبی موش نشان داده شد و تعامل مستقیم بین phospho-TAU و NDP52 توسط آزمایش های همبستگی ایمنی در موش و نمونه AD نشان داده شد ، با اشاره به نقش آن در تخریب TAU. جالب اینجاست که خاموش کردن NDP52 ، p62 یا NRF2 در سلولهای عصبی منجر به افزایش فسفو-TAU [113] ، [118] می شود. علاوه بر این ، افزایش دانه های APP داخل مغزی در هیپوکامپ موش های APP / PS1؟ E9 هنگامی که NRF2 وجود نداشت پیدا شد. این با نشانگرهای اتوفاژی تغییر یافته ، از جمله افزایش نسبتهای phospho-mTOR / mTOR و phospho-p70S6k / p70S6k (نشانگر مهار اتوفاژی) ، سطح افزایش یافته پیش کاتپسین D و تعداد بیشتری از اجسام چند لخته ای است [119]. در موش های بیان کننده APP انسانی (V717I) و TAU (P301L) ، کمبود NRF2 منجر به افزایش سطح کل و فسفاتو TAU در بخش نامحلول و افزایش تجمع APP داخل مغزی می شود ، همراه با کاهش سطح عصبی p62 ، NDP52 ، ULK1 ، ATG5 و GABARAPL1. هم محلی سازی بین پروتئین آداپتور p62 و APP یا TAU در غیاب NRF2 کاهش یافت [36]. به طور کلی ، این نتایج اهمیت NRF2 در اتوفاژی عصبی را برجسته می کند.
قوانین رونویسی مختلف به طور هماهنگ برای مداخله پروتواستازی
در شرایط حالت پایدار، پروتئاز با کنترل متابولیسم پروتئین-پروتئین و تغییرات پس از ترجمه، به دست می آید که پاسخ سریع را دریافت می کند. با این وجود، انطباق سلولی نیاز به تنظیم رونویسی UPR، UPS و ژنهای Autophagy دارد. با توجه به اینکه سلول های عصبی به طور مداوم به سوء هاضمه های سمی پایین، از جمله استرس اکسیداتیو و پروتوتوکسیک منتقل می شوند، تقویت پروتئازاز ناشی از مدولاسیون رونویسی می تواند به جلوگیری از انقراض مغز کمک کند.
در مورد UPR، فعال شدن هر یک از سه بازو در نهایت منجر به القاء رونویسی برخی ژن ها خواهد شد (در [43] بررسی شده است). به عنوان مثال، یک قطعه مشتق شده از ATF6 (ATF6f) به عناصر پاسخ ER استرس (ERSE) متصل می شود و باعث بیان چندین ژن از جمله XBPI، BIP و CHOP می شود. علاوه بر این، سیگنالینگ PERK موجب فعال شدن عامل رونویسی ATF4 می شود که بیان ژن های مربوط به چندین UPR را کنترل می کند و برخی دیگر از جمله ژنهای هدف NRF2 Hmox1 و p62 را کنترل می کنند. در نهایت، فعال سازی IRE1 منجر به تولید فاکتور رونویسی فعال، XBP1 (XBP1s) می شود که رونویسی ژن هایی را که پروتئین های رمزگذاری شده پروتئین را تشکیل می دهند، کنترل می کند.
از سوی دیگر، NRF1 برای بیان ژن پروتئازومایک در مغز ضروری است، زیرا موشهای Nrf1 ناخوشایند بیان ژن هایی را که با فرکانس های مختلف هسته 20S رمزگذاری می کنند، و همچنین مجموعه تنظیم کننده 19S همراه با اختلال عملکرد پروتئازامال 90 ] هر دو NRF1 و NRF2 به توالی ARE در ناحیه پروموتورهای ژن هدفشان متصل می شوند، که نشان می دهد که فعالیت های رونویسی همپوشانی دارند، هرچند که در مکانیسم های تنظیم کننده و مکان سازی سلولی [120] متفاوت است.
فاکتورهای رونویسی از جعبه Forkhead O (FOXO) خانواده کنترل بیان ژن های مربوط به چندین autophagy را کنترل می کند. همانند آنچه که با NRF2 اتفاق می افتد، چندین لایه تنظیم فعالیت های اعضای FOXO وجود دارد که می تواند بر استرس تغذیه ای یا اکسیداتیو [121] ایجاد شود. در نهایت، فاکتور رونویسی TFEB، به عنوان رگولاتور اصلی بیوگنسیس لیزوزومه در نظر گرفته شده است، نقش مهمی در تنظیم تنفس در شرایط تنش تغذیه ایفا می کند. بنابراين مهار mTORC1 منجر به انتقال هسته TFEB و القاء بيان ژنهاي اتوفاژي [122] مي شود.
در مجموع، وجود رگولاتورهای مختلف رونویسی این ماشین آلات، نشان می دهد که تداخل و مکانیسم هایی که به طور جزئی از کار بیفتند، ممکن است پروتوواستاز را تحت شرایط مختلف اطمینان دهند. بر این اساس، NRF2 ممکن است نقش قابل توجهی در بافت هایی داشته باشد که سطوح بالای استرس اکسیداتیو را پشتیبانی می کنند. به عنوان مثال، NRF2 ناشی از استرس اکسیداتیو ممکن است تحت شرایط غنی از مواد مغذی عمل کند تا رونوشت ریزی autofagy را تنظیم کند، شبیه آنچه که برای TFEB در شرایط گرسنگی یافت شده است. علاوه بر این، مغز عمدتا تحت شرایط غنی از مواد مغذی عمل می کند و NRF2 را به عنوان یک مکانیسم مناسب برای فعال شدن autophagy در نورون نشان می دهد.
نویدبخش پتانسیل درمانی NRF2 در پروتئینوپاتی ها
در چند سال گذشته، پیشرفت بزرگی در شناخت نقش های نظارتی UPR، UPS و Autophagy در فعالیت NRF2 و همچنین رونویسی متقابل NRF2 از اجزاء این سه سیستم انجام شده است. بنابراين، امكانات جديد درماني ممكن است بر اساس استثمار NRF2 به عنوان يك تنظيم كننده مهم ترين پروتكل كليه بيماري هاي نورولوژيكي ناشي شود.
با این حال ، یک سوال اساسی باقی مانده این است که آیا افزایش سطح NRF2 در مغز مفید یا مضر است. تجزیه و تحلیل داده های اپیدمیولوژیک ممکن است یک پاسخ جزئی ارائه دهد ، زیرا نشان می دهد که ژن NFE2L2 بسیار چندشکلی است و برخی از چند شکلی های نوکلئوتیدی منفرد موجود در منطقه تنظیم کننده پروموتر آن ممکن است طیف وسیعی از تنوع فیزیولوژیکی در بیان ژن در سطح جمعیت و برخی از هاپلوتیپ ها را فراهم کند. با کاهش خطر و / یا تاخیر در شروع AD ، PD یا ALS همراه بودند [123]. علاوه بر این ، همانطور که توسط هیز و همکارانش [124] مورد بحث قرار گرفت ، اثر NRF2 ممکن است پاسخی U شکل داشته باشد ، به این معنی که سطح NRF2 بسیار کم ممکن است منجر به از دست دادن محافظت از سلول و افزایش حساسیت به عوامل استرس زا شود ، در حالی که NRF2 زیاد ممکن است تعادل هموستاتیک را نسبت به یک سناریوی کاهشی (استرس کاهشی) ، که به غلط کشیدن و تجمع پروتئین کمک می کند. سطح NRF2 پایین در مغز این ایده را تأیید می کند که تنظیم جزئی جزئی برای دستیابی به منفی در شرایط پاتولوژیک کافی است. در حقیقت ، نقش محافظتی فعال سازی پاکسازی پروتئین با واسطه NRF2 در فرهنگ سلول های مختلف تخریب عصب و مدل های داخل بدن نشان داده شده است.
SFN یک فعال کننده دارویی NRF2 است که برای القای بیان ژن پروتئازومی و اتوفاژی نشان داده شده است [95] ، [36]. جالب توجه است که جو و همکارانش نشان دادند که SFN سطح TAU فسفریله را کاهش می دهد و Beclin-1 و LC3-II را افزایش می دهد ، که نشان می دهد فعال سازی NRF2 ممکن است تخریب این پروتئین سمی را از طریق اتوفاژی تسهیل کند [113]. علاوه بر این ، تخریب mHtt با SFN افزایش یافت و این با استفاده از MG132 برگردانده شد ، که نشانگر تخریب پروتئازومی این پروتئین سمی است [95]. تخریب اتوفاژی با واسطه فسفو و TAU نامحلول با فلاستین آلی fisetin گزارش شد. این ترکیب با ترویج همزمان فعال سازی و انتقال هسته ای هر دو TFEB و NRF2 ، همراه با برخی از ژن های هدف خود ، توانست autophagy را القا کند. این پاسخ با خاموش کردن TFEB یا NRF2 جلوگیری شد [125]. بوت و همکارانش اثرات مفیدی از یک فعال کننده همزمان NRF2 ، NRF1 و HSF1 بر مسمومیت پروتئین در آتروفی عضلانی نخاع و پیازچه ، یک اختلال تخریب عصبی ناشی از گسترش تکرارهای CAG کد کننده پلی گلوتامین که در آن مواد پروتئینی وجود دارد ، گزارش کردند [126]. پتانسیل فعال سازی NRF2 برای درمان اختلالات نورودژنراتیو با تأیید BG-12 ، فرمول دهانی دی متیل فومارات القا کننده NRF2 (DMF) ، برای درمان بیماری مولتیپل اسکلروزیس نشان داده شده است [127] ، [128]. موفقیت DMF در بیماری های خود ایمنی با یک جز inflammatory التهابی قوی نشان می دهد که بیماری های تخریب کننده عصبی ممکن است از جایگذاری مجدد این دارو سود ببرند. در یک مطالعه بالینی اخیر بر روی یک مدل؟ -sinucleinopathy PD ، DMF محافظت عصبی نشان داد ، که تا حدی ناشی از القای اتوفاژی است [129]. مطالعات گزارش شده در مورد اثرات مفید NRF2 در تخریب عصب اما تمرکز آن بر تأثیر آن بر ترشح پروتئین حتی بیشتر است (برای یک بررسی جامع ، مراجعه کنید به [7]). این کاملاً مرتبط است ، زیرا فرآیندهای آسیب رساننده متعددی را که می تواند همزمان با یک ضربه در NRF2 هدف قرار گیرد ، برجسته می کند ، همچنین شامل استرس اکسیداتیو ، التهاب عصبی یا اختلال عملکرد میتوکندری. با این حال ، برای تعیین اینکه آیا فعال سازی دارویی NRF2 ممکن است یک استراتژی معتبر برای تسهیل تخریب پروتئین های سمی در مغز باشد ، کارهای آینده مورد نیاز خواهد بود.
همانطور که قبلا توضیح داده شد ، GSK-3 بدتر می شود؟ فعالیت در بیماری های تخریب عصبی گزارش شده است و حدس زده شده است که در نتیجه کاهش NRF2 می تواند تا حدی مسئول نتیجه مخرب باشد. تحت این شرایط پاتولوژیک ، مهار کننده های GSK-3 همچنین می توانند برای افزایش سطح NRF2 و پروتئازاز همکاری کنند. اثرات مفید مهارکننده های GSK-3 در مدل های مختلف تخریب عصبی گزارش شده است و جالب تر ، سرکوب GSK-3 برای کاهش سطح پروتئین های سمی نشان داده شده است [130] ، [131] ، [132] ، [133]. اگرچه هنوز هیچ ارتباط مستقیمی بین مهار GSK-3 و تنظیم رونویسی NRF2 در ژنهای تقویت کننده پروتئوز مشاهده نشده است ، اما منطقی است که حدس بزنیم که تنظیم کم فعالیت GSK-3 منجر به افزایش سطح NRF2 می شود که در نهایت منجر به تقویت می شود. پروتئوستاز
فعالیت رونویسی NRF2 و همچنین ظرفیت سلولی برای حفظ پروتئوستاز با افزایش سن ، عامل اصلی خطر در ایجاد بیماری های نورودژنراسیون ، کاهش می یابد. منطقی است که فکر کنیم تقویت NRF2 و در نتیجه پروتئوستاز حداقل باعث جمع شدن تجمع مواد پروتئینی و تخریب عصبی می شود. در واقع ، درمان فیبروبلاست های پیر انسان با تری ترپنوئید 18 اسید-گلیسیرتینیک اسید (18؟ -GA) باعث فعال شدن NRF2 شده و منجر به القای پروتئازوم و افزایش طول عمر می شود. این مطالعه نشان می دهد که فعال سازی دارویی NRF2 حتی در اواخر عمر نیز امکان پذیر است [86]. علاوه بر این ، یک مطالعه بعدی نشان داد که این ترکیب با واسطه SKN-1 و فعال سازی پروتئازوم در C.elegans با اثرات مفید بر پیشرفت AD در مدل های نماتد مربوطه [134].
همه چیز در نظر گرفته شده است، القا ژن وابسته به پروتئازوز توسط NRF2 منجر می شود مفید در پروتئینوپاتی های مختلف.
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
فاکتور هسته ای 2 (NF-E2) حاصل از 2 (NF-E2) وابسته به عامل XNUMX، در غیر این صورت به عنوان NrfXNUMX شناخته شده، فاکتور رونویسی است که بیان کننده آنزیم های مختلف آنتی اکسیدان و سم زدایی آن را تنظیم می کند. تحقیقات انجام شده نقش خود را در کنترل استرس اکسیداتیو نیز نشان داده اند. اکثر بیماری های نورولوژیک مانند بیماری آلزایمر و بیماری پارکینسون با استرس اکسیداتیو و التهاب مزمن مشخص می شوند، اهداف مشترک روشهای درمان Nrf2. دکتر الکس جیمنز DC، CCST Insight
اظهارات نهایی
فاکتور رونویسی NRF2 یک پاسخ پروتئستاتیک را با حساسیت به تغییرات و تغییرات در UPR، UPS و autophagy (شکل 4) ارزیابی می کند. در نتیجه، کمبود NRF2 نشان داده است که پروتئینوپتیک را تشدید می کند، و این نشان می دهد که NRF2 برای ترخیص پروتئین مطلوب ضروری است. همه با هم، ما می توانیم حدس بزنیم که NRF2 ممکن است هدف درمان جالب برای پروتئینوپاتی است.
شکل 4 NRF2 به عنوان یک هاب اتصال سیگنال های اضطراری حاصل از پروتو توکسین به یک واکنش رونویسی محافظتی. انباشت پروتئین های باز / غلط شده منجر به فعال شدن پاسخ پروتئین باز (UPR) در ER می شود. فعال سازی PERK یا MAPK ممکن است القاء رونویسی Gpx8 ساکن ER و چندین آنزیم تنظیم کننده سطح GSH را داشته باشد که برای اطمینان از تاول زدن پروتئین صحیح ضروری است. میله های پروتئینی از فعالیت پروتئازوما (UPS) جلوگیری می کنند، احتمالا از تخریب NRF2 اجتناب می کنند. NRF2 نشان داده شده است که به طور خاص رونویسی ژن Psma3، Psma6، Psmb1، Psmb5 و Pomp را تنظیم می کند. چندین واحد دیگر به صورت وابسته به NRF2 در پاسخ به D3T تنظیم شد، احتمالا لیستی از زیرمجموعه های پروتئازوم تنظیم شده توسط NRF2 را افزایش داد. Autophagy راه اصلی برای تخریب پروتئین است. اتوفاژی را نیز تنظیم می NRF2، اتصال این مسیر تخریب با NRF2 القایی رونویسی از p62، Ndp52، Ulk1، Atg2b، Atg4c، Atg5، Atg7 و Gabarapl1.
طبق مقاله فوق، در حالی که علائم بیماری های نورودنراتیو را می توان از طریق انواع گزینه های درمان درمان کرد، مطالعات انجام شده نشان داده است که فعال شدن Nrf2 می تواند یک روش درمان مناسب باشد. زیرا Activators Nrf2 مکانیسم های بیماری را هدف قرار می دهند، تمام بیماری های تخریب عصبی می توانند از استفاده از فاکتور رونویسی Nrf2 بهره مند شوند. یافته های Nrf2 انقلابی در درمان بیماری های تخریب عصبی ایجاد کرده است. دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
زانو درد یک علامت شناخته شده است که می تواند به دلیل آسیب دیدگی های مختلف زانو و / یا شرایط ، از جمله رخ دهدآسیب های ورزشی. زانو یکی از پیچیده ترین مفاصل در بدن انسان است زیرا از تقاطع چهار استخوان ، چهار رباط ، تاندون های مختلف ، دو مینیسک و غضروف تشکیل شده است. طبق آكادمی پزشكان خانواده آمریكا ، شایع ترین دلایل درد زانو شامل زیر گرفتگی كشكك ، تاندینیت استخوان کشکک یا زانوی جهنده و بیماری اوسگود - شلاتر است. اگرچه زانو درد به احتمال زیاد در افراد بالای 60 سال بروز می کند ، اما درد زانو می تواند در کودکان و نوجوانان نیز رخ دهد. درد زانو را می توان با استفاده از روش های RICE در خانه درمان کرد ، با این حال ، آسیب های شدید زانو ممکن است نیاز به مراقبت فوری پزشکی ، از جمله مراقبت های کایروپراکتیک داشته باشد.
استرس اکسیداتیو به عنوان آسیب سلولی ناشی از رادیکال های آزاد یا مولکول های ناپایدار که در نهایت بر عملکرد سالم تاثیر می گذارد، توصیف می شود. بدن انسان رادیکال های آزاد را برای خنثی کردن باکتری ها و ویروس ها ایجاد می کند، اما عوامل خارجی مانند اکسیژن، آلودگی و اشعه می توانند اغلب رادیکال های آزاد تولید کنند. استرس اکسیداتیو با مسائل بهداشتی متعددی مرتبط است.
استرس اکسیداتیو و سایر عوامل استرس زائی مکانیسم های محافظتی داخلی است که می تواند به تنظیم پاسخ آنتی اکسیدانی بدن انسان کمک کند. Nrf2 پروتئینی است که سطوح استرس اکسیداتیو را حس می کند و سلول ها را قادر می سازد خود را از عوامل داخلی و خارجی محافظت کند. Nrf2 نیز برای کمک به تنظیم ژن های درگیر در تولید آنزیم های آنتی اکسیدان و ژن های استرس واکنش نشان داده شده است. هدف مقاله زیر این است که توضیح دهد اثرات Nrf2 در سرطان
چکیده
مسیر Keap1-Nrf2 تنظیم کننده اصلی پاسخ های سیتوپروتئینی به استرس اکسیداتیو و الکتروفلی است. اگر چه مسیرهای سیگنالینگ سلولی که توسط فاکتور رونویسی Nrf2 منجر به جلوگیری از ابتلا به سرطان و پیشرفت در بافت های نرمال و زودرس می شود، در سلول های کاملا بدخیم فعالیتی Nrf2 با افزایش مقاومت شیمیایی سلولی و افزایش رشد سلول های تومور باعث افزایش رشد می شود. در این بررسی گرافیکی، ما یک مرور کلی از مسیر Keap1-Nrf2 و تنظیم غلط آن در سلول های سرطانی ارائه می دهیم. ما همچنین خلاصه ای از عواقب فعال سازی قانونی Nrf2 در سلول های سرطانی را خلاصه می کنیم و چگونه می توان در ژن درمان سرطان مورد بهره برداری قرار گرفت.
مسیر Keap1-Nrf2 تنظیم کننده اصلی پاسخ های محافظ سلولی به تنش های درون زا و اگزوژن ناشی از گونه های فعال اکسیژن (ROS) و الکتروفیل ها است [1]. پروتئینهای سیگنالدهنده کلیدی در مسیر عبارتند از فاکتور رونویسی Nrf2 (فاکتور 2 مربوط به فاکتور هستهای اریتروئید 2) که با پروتئینهای کوچک Maf به عنصر پاسخ آنتیاکسیدانی (ARE) در مناطق تنظیمکننده ژنهای هدف متصل میشود و Keap1 (Kelch ECH) پروتئین مرتبط 1)، یک پروتئین سرکوب کننده است که به Nrf2 متصل می شود و باعث تخریب آن توسط مسیر پروتئازوم یوبیکوئیتین می شود (شکل 1). Keap1 یک پروتئین بسیار غنی از سیستئین است، Keap1 موش در مجموع دارای 25 و انسان 27 باقیمانده سیستئین است که اکثر آنها می توانند در شرایط آزمایشگاهی توسط اکسیدان ها و الکتروفیل های مختلف اصلاح شوند [2]. سه مورد از این باقیماندهها، C151، C273 و C288، با تغییر ساختار Keap1 که منجر به جابجایی هستهای Nrf2 و متعاقب آن بیان ژن هدف میشود، نقش عملکردی دارند (شکل 3). مکانیسم دقیقی که به موجب آن تغییرات سیستئین در Keap1 منجر به فعالسازی Nrf1 میشود، مشخص نیست، اما دو مدل غالب اما متقابلاً منحصربفرد نیستند (2) مدل «لولا و چفت»، که در آن تغییرات Keap1 در باقیماندههای تیول موجود در IVR Keap1 است. برهمکنش با Nrf1 را مختل میکند و باعث ایجاد ناهماهنگی باقیماندههای لیزین در Nrf2 میشود که دیگر نمیتوان پلیابیکوئیتینیله کرد و (2) مدلی که در آن اصلاح تیول باعث تفکیک Cul2 از Keap3 میشود [1]. در هر دو مدل، Keap3 اصلاحشده با القاء و Nrf2 غیرفعال میشود و در نتیجه پروتئینهای Nrf1 جدید سنتز شده Keap2 را دور زده و به هسته منتقل میشوند، به ARE متصل میشوند و بیان ژنهای هدف Nrf1 مانند NAD(P)H را هدایت میکنند. کینون اکسیدوردوکتاز 2 (NQO1)، هم اکسیژناز 1 (HMOX1)، گلوتامات سیستئین لیگاز (GCL) و گلوتاتیون S ترانسفرازها (GSTs) (شکل 1). علاوه بر تغییرات تیول های Keap2 که منجر به القای ژن هدف Nrf1 می شود، پروتئین هایی مانند p2 و p21 می توانند به Nrf62 یا Keap2 متصل شوند و در نتیجه تعامل بین Nrf1 و Keap2 را مختل کنند [1]، [1] (شکل 3).
شکل 1. ساختارهای Nrf2 و Keap1 و کد سیستئین. (الف) Nrf2 متشکل از 589 اسید آمینه و دارای شش حوزه تکاملی بسیار حفاظت شده، Neh1-6. Neh1 حاوی یک موتیف bZip، یک ساختار ناحیه پایه زیپ لوسین (L-Zip) است، که در آن ناحیه پایه مسئول تشخیص DNA است و L-Zip واسطه دیمر شدن با پروتئین های کوچک Maf است. Neh6 به عنوان یک دگرون برای واسطه تخریب Nrf2 در هسته عمل می کند. Neh4 و 5 دامنه های ترانفعال سازی هستند. Neh2 حاوی نقوش ETGE و DLG است که برای برهمکنش با Keap1 مورد نیاز است، و یک ناحیه آبدوست از باقیماندههای لیزین (7 K) که برای پلی یوبی کوئیتیناسیون وابسته به Keap1 و تخریب Nrf2 ضروری هستند. (ب) Keap1 از 624 باقیمانده اسید آمینه تشکیل شده است و دارای پنج دامنه است. دو موتیف برهمکنش پروتئین و پروتئین، دامنه BTB و دامنه Kelch، توسط ناحیه مداخله گر (IVR) از هم جدا می شوند. دامنه BTB همراه با بخش N ترمینال IVR واسطه همودایمریزاسیون Keap1 و اتصال با Cullin3 (Cul3) است. دامنه کلچ و ناحیه ترمینال C واسطه تعامل با Neh2 هستند. (C) Nrf2 از طریق نقوش Neh1 ETGE و DLG با دو مولکول Keap2 تعامل دارد. هر دو ETGE و DLG به سایت های مشابه در سطح پایین موتیف Keap1 Kelch متصل می شوند. (د) Keap1 غنی از باقی مانده های سیستئین است، با 27 سیستئین در پروتئین انسانی. برخی از این سیستئین ها در نزدیکی باقی مانده های اساسی قرار دارند و بنابراین اهداف عالی الکتروفیل ها و اکسیدان ها هستند. الگوی اصلاح باقی مانده های سیستئین توسط الکتروفیل ها به عنوان کد سیستئین شناخته می شود. فرضیه کد سیستئین پیشنهاد میکند که عوامل فعالکننده Nrf2 از نظر ساختاری بر سیستئینهای مختلف Keap1 تأثیر میگذارند. تغییرات سیستئین منجر به تغییرات ساختاری در Keap1 می شود که تعامل بین دامنه های Nrf2 DLG و Keap1 Kelch را مختل می کند، بنابراین پلی یوبی کوئیتیناسیون Nrf2 را مهار می کند. اهمیت عملکردی Cys151، Cys273 و Cys288 نشان داده شده است، زیرا Cys273 و Cys288 برای سرکوب Nrf2 و Cys151 برای فعال سازی Nrf2 توسط القاء مورد نیاز هستند [1]، [3].
شکل 2 مسیر سیگنالینگ Nrf2-Keap1. (A و B) در شرایط پایه، دو مولکول Keap1 به Nrf2 پیوند می دهند و Nrf2 به وسیله مجتمع لیزاز E3 مبتنی بر Cul3 Polyubiquitylated است. این Polyubiquitilation منجر به تجزیه سریع Nrf2 توسط پروتئازوم. بخش کوچکی از Nrf2 از ترکيب مهار کننده فرار می کند و در هسته تجمع می يابد تا بتواند بيان ژن وابسته به ARE پايه ايجاد کند، به اين ترتيب هوموستاز سلولی را حفظ می کند. (C) تحت شرایط تنش، القا کننده ها، کیت های Keap1 را منعکس می کنند که منجر به مهار بیوفیزیکسیون Nitf2 از طریق جداسازی مجتمع مهار کننده می شود. (D) با توجه به مدل لولا و لنگ، اصلاح بقایای سیکستی Keap1 خاص منجر به تغییرات کنترلی در Keap1 می شود که منجر به جدا شدن موتیف Nrf2 DLG از Keap1 می شود. مسدود سازی Nrf2 مختل می شود اما اتصال با موتیف ETGE باقی می ماند. (E) در مدل جداسازی Keap1-Cul3، اتصال به Keap1 و Cul3 در پاسخ به الکتروفیل ها مختل می شود، که منجر به فرار از Nrf2 از سیستم ubiquitination می شود. در هر دو مدل پیشنهاد، القاء اصلاح شده و Nrf2 محدود Keap1 غیر فعال است و در نتیجه، پروتئین Nrf2 تازه سنتز دور زدن Keap1 و جابجا کردن درون هسته، اتصال به واکنش عنصر آنتی اکسیدان (ARE) و رانندگی بیان هدف Nrf2 ژن هایی مانند NQO1، HMOX1، GCL و GSTs [1]، [3].
شکل 3 مکانیسم برای تجمع هسته ای هسته ای Nrf2 در سرطان. (A) جهش های اجتماعی در Nrf2 یا Keap1 باعث اختلال در تعامل این دو پروتئین می شود. در Nrf2، جهش های موشی ETGE و DLG را تحت تاثیر قرار می دهند، اما در جهش های Keap1 بیشتر توزیع می شوند. علاوه بر این، فعال سازی آن کوهن مانند KrasG12D [5]، یا اختلال مهار کننده های توموری مانند PTEN [11] می تواند منجر به رونویسی القاء Nrf2 و افزایش هسته Nrf2 شود. (B) Hypermethylation پروپران Keap1 در سرطان ریه و پروستات منجر به کاهش بیان mRNA Keap1، که تجمع هسته ای Nrf2 [6]، [7] را افزایش می دهد. (C) در کارسینومای پاپیلری کلیوی، از دست دادن فعالیت آنزیم هیدراتاز فومارات منجر به انباشت فومارات و به سویکسیون شدن پسماندهای سیتین Keap1 (2SC) می شود. این اصلاح post-translational منجر به اختلال در تعامل Keap1-Nrf2 و تجمع هستهی Nrf2 [8]، [9] می شود. (D) انباشت پروتئین های تخریب کننده مانند p62 و p21 می تواند باعث اتصال Nof2-Keap1 شود و باعث افزایش تعداد هسته Nrf2 می شود. p62 به Keap1 متصل می شود، همپوشانی جیب اتصال برای Nrf2 و p21 به طور مستقیم با DLG و ETGE های motifs Nrf2 تعامل دارد، در نتیجه با Keap1 [10] رقابت می کند.
مکانیسم فعال سازی و تنظیم مقررات Nrf2 در سرطان
گرچه سیتوپراکسیتی که توسط فعال سازی Nrf2 ارائه می شود برای پیشگیری شیمیایی در سرطان ها در بافت های طبیعی و زودرس مهم است، در فعالیت سلول های کاملا بدخیم فعالیتی Nrf2 با افزایش مقاومت به شیمیدرمانی و افزایش رشد سلول های تومور [4] مزایای رشد را فراهم می آورد. چندین مکانیزم که توسط مسیریابی سیگنال nf2 فعالانه در سرطان های مختلف فعال شده اند، شرح داده شده است: (جهش های Somatic 1) در Keap1 یا ناحیه اتصال پیوند Keap1 از Nrf2 باعث اختلال در تعامل آنها می شود. (2) خاموش کردن اپی ژنتیک بیان Keap1 منجر به سرکوب نامناسب Nrf2؛ (3) انباشت پروتئین های تخریب کننده مانند p62 منجر به جدایی از مجموعه Keap1-Nrf2؛ (4) القاء رونویسی Nrf2 به وسیله Oncogenic K-Ras، B-Raf و c-Myc؛ و (5) اصلاح پس از ترجمه کیپسینومکس توسط سکتسی سیلسیون که در کارسینومای فامارات هیدراتاز [1]، [3]، [4]، [5]، [6]، [7]، [8]، [9]، [10] 3]، [2]، [2] (شکل 4). غنی پروتئین Nrf2 غالبا باعث افزایش بیان ژن های متابولیسم دارو می شود که باعث افزایش مقاومت به داروهای شیمی درمانی و پرتودرمانی می شود. علاوه بر این، سطح پروتئین بالا Nrf11 با پیش آگهی ضعیف در سرطان مرتبط است [4]. Overfacial NrfXNUMX همچنین با تکثیر گلوکز و گلوتامین به سمت مسیرهای آنابولیک، به افزایش سنتز پورین و تأثیر آن بر مسیر پنتوز فسفات برای ترویج تکثیر سلولی [XNUMX] (شکل XNUMX) اثر می گذارد.
شکل 4 نقش دوگانه Nrf2 در توموری زایی. در شرایط فیزیولوژیکی، سطح پایین هسته Nrf2 برای نگهداری هوموتازای سلولی کافی است. Nrf2 مهار آغاز تومور و متاستاز سرطان را با حذف مواد سرطانزا، ROS و دیگر عوامل آسیب رساندن به DNA متوقف می کند. در طی tumorigenesis، تجمع آسیب DNA منجر به افزایش بیش فعالی از Nrf2 است که به سلول های بدخیم مستقل برای تحمل سطوح بالای ROS درونی و جلوگیری از آپوپتوز کمک می کند. سطوح پایدار هستی Nrf2، علاوه بر ژنهای سیتوپروتئینی که به برنامه ریزی متابولیکی و افزایش تکثیر سلولی کمک می کند، ژنهای متابولیک را فعال می کنند. سرطان هایی با سطح Nof2 بالایی با پیش آگهی ضعیف همراه هستند زیرا از طریق رادیو و شیمیایی مقاومت و تکثیر سلول های سرطانی تهاجمی. بنابراین فعالیت مسیری Nrf2 در مراحل اولیه تومور زایی محافظ است، اما در مراحل بعدی آسیب می بیند. بنابراین، برای جلوگیری از سرطان، افزایش فعالیت nof2 همچنان یک رویکرد مهم است، در حالیکه برای درمان سرطان، مهار نفی2 مطلوب است [4]، [11].
با توجه به اینکه فعالیت بالا Nrf2 در سلول های سرطانی با نتایج نامطلوب معمولا رخ می دهد، نیازی به درمان هایی برای مهار کردن Nrf2 وجود دارد. متأسفانه، با توجه به شباهت ساختاری با برخی از اعضای خانواده bZip، توسعه مهارکننده های خاص Nrf2 یک کار چالش برانگیز است و تنها چند مطالعۀ مهار نفیسه نیومکس تا به امروز منتشر شده است. با نمایش محصولات طبیعی، Ren et al. [2] ترکیبی از بروساتول را به عنوان یک مهارکننده Nrf12 شناسایی کرد که اثربخشی شیمی درمانی سیس پلاتین را افزایش می دهد. علاوه بر این، مهارکننده های PI2K [3]، [11] و Nrf13 siRNA [2] برای مهار Nrf14 در سلول های سرطانی مورد استفاده قرار گرفته اند. اخیرا، ما یک روش جایگزین، شناخته شده به عنوان ژن درمانی سرطانی سرطانی، برای هدف قرار دادن سلول های سرطانی با سطوح بالای Nof2 مورد استفاده قرار داده ایم. بردارهای lentiviral [xnumx] حاوی تیمیدین کیناز (TK) حاوی Nrf2 به سلول های سرطانی با فعالیت ARE بالا منتقل می شوند و سلول ها با یک پروتیین، ganciclovir (GCV) درمان می شوند. GCV به GCV-monophosphate متابولیزه می شود که به وسیله کیناز سلولی به فرم تری فسفات سمی [2] (شکل 15) فسفریل می شود. این باعث مرگ موثر نه تنها سلول های تومور TK، بلکه سلول های همسایه به علت اثر متقابل [16] می شود. ژن درمان TK / GCV تحت کنترل ARE را می توان از طریق ترکیب دیکسروبوسیین عامل chemotherapeutic agent سرطان به درمان [5]، حمایت از این تصور که این رویکرد می تواند در ارتباط با درمان های سنتی مفید باشد.
شکل 5 درمان ژن خودکشی. تجمع هستهی Nrf2 مؤثر در سلولهای سرطانی می تواند با استفاده از بردار ویروسی راننده Nrf2 برای ژن درمانی سرطانی سرطانی [16] مورد سوء استفاده قرار گیرد. در این روش، ویروس lentiviral (LV) بیان کننده تیمیدین کیناز (TK) تحت حداقل پروتئور SV40 با چهار ARE ها به سلول های آدنوکارسینوم ریه منتقل می شود. سطوح هسته ای Nrf2 منجر به بیان قوی TK از طریق اتصال به Nrf2 می شود. سپس سلولها با داروهای پیشگیرانه، گانسیکلوویر (GCV) درمان می شوند که توسط TK فسفریل می شود. GCV Triphosphorylated خنثی سازی DNA را ایجاد می کند و منجر به کشتن موثر سلولهای توموری نه تنها TK، بلکه همچنین سلول های همسایه به علت اثرات جانشین.
Nrf2 یک تنظیم کننده اصلی است که باعث تولید آنتی اکسیدان های قوی در بدن انسان می شود که به کاهش استرس اکسیداتیو کمک می کند. آنزیم های آنتی اکسیدان مختلف مانند سوپراکسید دیسموتاز یا SOD، گلوتاتیون و کاتالاز نیز از مسیر Nof2 فعال می شوند. علاوه بر این، بعضی از مواد شیمیایی فیتوکمیک مانند زردچوبه، آسیوآنگاندا، بوکوپا، چای سبز و خار، Activate Nrf2 را فعال می کنند. مطالعات تحقیقاتی آن را یافته اند فعال سازی Nrf2 می تواند به طور طبیعی محافظت سلولی را افزایش دهد و تعادل را به بدن انسان بازگرداند.
دکتر الکس جیمنز DC، CCST Insight
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
تشکر و قدردانی
این کار توسط آکادمی فنلاند، بنیاد سیگرید جوسلیوس و سازمان های سرطان فنلاند پشتیبانی شد.
در نتیجه، فاکتور هسته ای (2 مشتق از اریتروئید) مانند 2، همچنین به عنوان NFE2L2 یا Nrf2 شناخته می شود، پروتئینی است که تولید آنتی اکسیدان هایی را افزایش می دهد که از بدن انسان در برابر استرس اکسیداتیو محافظت می کند. همانطور که در بالا توضیح داده شد، تحریک مسیر Nrf2 برای درمان بیماری های ناشی از استرس اکسیداتیو، از جمله سرطان، در حال بررسی است. دامنه اطلاعات ما محدود به مسائل مربوط به کایروپراکتیک و سلامت ستون فقرات است. برای بحث در مورد موضوع، لطفاً از دکتر خیمنز بپرسید یا با ما تماس بگیرید915-850-0900.
زانو درد یک علامت شناخته شده است که می تواند به دلیل آسیب دیدگی های مختلف زانو و / یا شرایط ، از جمله رخ دهدآسیب های ورزشی. زانو یکی از پیچیده ترین اتصالات بدن انسان است که از تقاطع چهار استخوان، چهار رباط، تاندون های مختلف، دو منیسکی و غضروف ساخته شده است. به گفته آکادمی پزشکان خانواده آمریکایی، شایع ترین علل درد زانو شامل سوزش پاتلا لار، تاندونیت پاتلا و یا زانو بلوز و بیماری Osgood-Schlatter می باشد. گرچه درد زانو بیشتر در افراد بالای 60 رخ می دهد، درد زانو نیز در کودکان و نوجوانان رخ می دهد. بعد از روش های RICE، درد زانو را می توان در خانه درمان کرد، با این حال، آسیب های شدید زانو ممکن است نیاز به مراقبت فوری پزشکی، از جمله مراقبت از مراقبت از ماساژ.
DNA از حدود ژنهای 20,000 پشتیبانی می کند، هر کدام یک برنامه برای ایجاد یک پروتئین یا آنزیم مورد نیاز برای یک شیوه زندگی سالم دارد. هر یک از این الگوهای باید به طور مداوم توسط نوعی "پروموتر" تنظیم شود که دقیقا مقدار زیادی از هر ماده و / یا شیمیایی تولید می کند و تحت شرایطی نیز چنین خواهد شد.
با اتصال به یک نوع خاص از پروموتر سوئیچ مانند، که به عنوان عنصر پاسخ آنتی اکسیدان شناخته می شود، یا ARE، فاکتور nrf2hundredsاز سرعت ایجاد صدها ژن مجزا پشتیبانی می کند که سلول ها را تحت شرایط استرس زا قادر به زنده ماندن می کند. این ژن ها سپس مجموعه ای از آنزیم های آنتی اکسیدان را تولید می کنند که با خنثی سازی اکسیدان ها و تمیز کردن مواد جانبی سمی باقی مانده در تولید ، علاوه بر کمک به بازگرداندن آسیب هایی که ایجاد می کنند ، یک شبکه دفاعی ایجاد می کنند.
استرس اکسیداتیو چیست؟
چندین اکسیدان مانند رادیکال سوپراکسید یا O2- و پراکسید هیدروژن یا H2O2 از طریق سوزاندن مواد و / یا مواد شیمیایی بدن انسان ایجاد شده است. بدن انسان دارای آنزیم های آنتی اکسیدان است که غذاها و نوشیدنی های واکنشی را که مصرف می کنیم ، خنثی و سم زدایی می کند. Nrf2 تولید خود را تعدیل می کند تا تعادل را حفظ کند و تقاضای همه این آنزیم ها را تأکید می کند. این تعادل را می توان با عوامل مختلفی از جمله سن قطع کرد.
با افزایش سن ، "بدن انسان Nrf2 کمتری ایجاد می کند و این تعادل ظریف می تواند به تدریج شروع به چرخش به سمت اکسیداتیو کند ، حالتی که از آن به عنوان استرس اکسیداتیو یاد می شود. بیماری همچنین ممکن است باعث تولید بیش از حد اکسیدان شود. عفونت ها ، آلرژی ها و اختلالات خود ایمنی می توانند سلول های ایمنی بدن ما را برای ایجاد اکسیدان های واکنشی مانند O2- تحریک کنند. ، H2O2 ، OH و HOCl ، جایی که سلولهای سالم آسیب دیده و با التهاب پاسخ می دهند. بیماری های مرتبط با پیری ، از جمله حملات قلبی ، سکته مغزی ، سرطان ، و شرایط تخریب عصبی مانند بیماری آلزایمر ، همچنین تولید اکسیدان ها را افزایش می دهد ، باعث ایجاد استرس و پاسخ التهاب می شود.
Activators Nrf2 چیست؟
پروتئین nrf2، همچنین به عنوان عامل فاکتور رونویسی شناخته شده به علت اینکه آنزیم ها و ژن ها را پشتیبانی و کنترل می کند، عنصر سری یک دنباله ای از واکنش های بیوشیمیایی در سلول است که به تغییرات در تعادل شناختی و همچنین تعادل اکسیداتیو واکنش نشان می دهد. عناصر حساس این مسیر تغییر و تخلیه Nof2، باعث آن می شود، بنابراین ممکن است به هسته سلول به سمت DNA گسترش یابد. Nrf2 ممکن است به طور متناوب، ژن ها و آنزیم هایی را که برای محافظت از سلول ها پشتیبانی می کند، روشن یا خاموش کند.
خوشبختانه، انواع مختلفی از مواد فعال کننده Nrf2 از طریق مصرف گیاهان و عصاره های خاصی که قرنها پیش در درمان های سنتی چینی و بومی آمریکایی استفاده می شوند، توسعه می یابد. به نظر می رسد این فیتوکمیکال ها به همان اندازه قدرتمند با عوارض جانبی کمتری هستند، مانند محصولات دارویی فعال Nrf2 که امروزه استفاده می شوند.
فاکتور هسته ای فاکتور اریتروتین 2، که معمولا به نام Nrf2 شناخته می شود، فاکتور رونویسی است که سلول را با تنظیم ژن ها، آنزیم ها و پاسخ های آنتی اکسیدان محافظت می کند. فاکتورهای رونویسی یک نوع پروتئینی است که به DNA برای ترویج ایجاد مواد خاص و مواد شیمیایی مانند گلوتاتیون S-transferases یا GST متصل می شود. فعال سازی Nrf2 باعث تولید پروتئین های فعال می شود که توانایی آنتی اکسیدان قوی را برای کاهش استرس اکسیداتیو نشان می دهد.
دکتر الکس جیمنز DC، CCST Insight
علم پس از Activation Nrf2
هنگامی که مکمل اولیه غذایی Nrf2 فعال در 2004 ایجاد شد، حداقل اطلاعات مربوط به عملکرد مسیر nrf2 شناخته شد. روزنامه های تقریبا 200 در ادبیات درباره Nrf2، همچنین به عنوان عامل هسته ای مانند 2 یا NFE2L2 شناخته می شوند، وجود داشت و محققان فقط شروع به کشف پاسخ آنتی اکسیدانی Nrf2 در پستانداران کردند. با این حال، از 2017، بیش از 9,300 پژوهش های علمی در مورد این "تنظیم کننده اصلی"، چاپ شده است.
در حقیقت، Nrf2 بسیاری از آنزیم های آنتی اکسیدان را تنظیم می کند که به ژن ها وابسته نیستند؛ در عوض، آنها را در برابر انواع مختلف شرایط مرتبط با استرس قرار می دهد که در سلول ها، اندام ها و در نهایت ارگانیسم ها در شرایط سالم و پاتولوژیک قرار می گیرند. بر اساس این اطلاعات جدید از مطالعات پژوهش علمی منتشر شده، محققان اکنون می توانند بهتر پیشرفت کنند مکمل های غذایی Nrf2.
از سال 2007 ، مطالعات research عملکرد پیچیده مسیر Nrf2 را نشان داده است. مشخص شده است که فعال کننده های Nrf2 از عوامل مختلف ساختارهای بدن انسان تقلید می کنند. از طریق این مسیرها ، فعال کننده های Nrf2 به منظور ایجاد تعادل و پاسخگویی به نیازهای در حال تکامل ژن ها ، مجهز شده اند تا شرایط متغیر را در سراسر سلول احساس کنند.
چرا از مکمل های فعال Nrf2 استفاده می کنید؟
همانطور که توانایی فعال شدن Nrf2 با سن در ارگانیسم ها کاهش می یابد، ممکن است تغییرات ایجاد شود. مطالعات انجام شده نشان داده است که تمرکز Nrf2 در سلول ها با سن کاهش می یابد و شاخص های افزایش استرس اکسیداتیو را نشان می دهد. انواع بیماری های مربوط به سن مانند آترواسکلروز و بیماری های قلبی عروقی، آرتریت، سرطان، چاقی، دیابت نوع 2، فشار خون بالا، آب مروارید، بیماری آلزایمر و همچنین بیماری های پارکینسون می توانند به دلیل این تغییرات ایجاد شوند. استرس اکسیداتیو با این مسائل بهداشتی یافت شده است.
با تحریک ظرفیت سلول برای افزایش تولید فعال کننده Nrf2، مکمل های غذایی Nrf2 می تواند به بهبود توانایی بدن انسان برای مقابله با اثرات استرس اکسیداتیو کمک کند. اسیدهای چرب اشباع نشده و یا PUFA ها یکی از مولکول های اکسیده شده به راحتی هستند و به خصوص برای آسیب رساندن به رادیکال های آزاد آسیب پذیرند. تولید اسید تیوباربیتوریک یا TBARS می تواند با افزایش سن افزایش یابد، که نشان دهنده استرس اکسیداتیو افزایش یافته همراه با کاهش جریان های کنترل شده Nrf2 می باشد.
از نظر زیست شناختی ، القای ژن مکانیزم واقعا آهسته ای است ، به طور کلی ساعت ها برای انتقال از طریق یک مسیر نیاز دارند. در نتیجه ، آنزیم های many سوئیچ های روشن / خاموش خود را دارند که می تواند در عرض چند دقیقه توسط آنزیم های تنظیم کننده مختلف ایجاد شود. محققان ترکیبات اختصاصی فعال کننده های Nrf2 را توسعه داده اند که از این پایگاه دانش فعال سازی استفاده می کنند. فعال سازی Nrf2 نه تنها از فاکتور رونویسی Nrf2 که از مهار کننده آن تخلیه شده و به هسته سلول مهاجرت می کند تشکیل شده است ، بلکه همچنین به توالی های DNA اختصاصی متصل می شود تا بیان ژن محافظت از سلول را تشویق کند ، تنظیم سرعت خارج شدن Nrf2 از هسته.
درک روش از بین بردن و فعال شدن Nrf2 در بدن انسان به محققان این امکان را داده است تا ترکیبی از فعال کننده های مختلف Nrf2 ایجاد کنند تا بازتاب ژن ها را از طریق مدولاسیون انجام دهند. ترکیبی از دانش بنیان ، همراه با طیف گسترده ای از سایر تحقیقات تحقیقاتی به تولید فعال کننده های Nrf2 برای استفاده به عنوان مکمل های غذایی کمک کرده است. دامنه اطلاعات ما به مسائل مربوط به بهداشت عمل جراحی و ستون فقرات محدود می شود. برای بحث در مورد موضوع ، لطفا در صورت تمایل از دکتر خیمنز بپرسید یا با ما تماس بگیرید915-850-0900.
دکتر الکس جیمنز سرپرستی می کند
بحث موضوعی اضافی: کاهش درد زانو بدون جراحی
زانو درد یک علامت شناخته شده است که می تواند به دلیل آسیب دیدگی های مختلف زانو و / یا شرایط ، از جمله رخ دهدآسیب های ورزشی. زانو یکی از پیچیده ترین اتصالات بدن انسان است که از تقاطع چهار استخوان، چهار رباط، تاندون های مختلف، دو منیسکی و غضروف ساخته شده است. به گفته آکادمی پزشکان خانواده آمریکایی، شایع ترین علل درد زانو شامل سوزش پاتلا لار، تاندونیت پاتلا و یا زانو بلوز و بیماری Osgood-Schlatter می باشد. گرچه درد زانو بیشتر در افراد بالای 60 رخ می دهد، درد زانو نیز در کودکان و نوجوانان رخ می دهد. بعد از روش های RICE، درد زانو را می توان در خانه درمان کرد، با این حال، آسیب های شدید زانو ممکن است نیاز به مراقبت فوری پزشکی، از جمله مراقبت از مراقبت از ماساژ.
آنتی اکسیدان ها به صورت علمی به عنوان ترکیباتی که فرایند اکسیداسیون را در بدن انسان محدود می کنند، به صورت علمی به ارمغان می آورند، که اگر باقی بمانند، می توانند رادیکال های آزاد ایجاد کنند که می توانند واکنشهای زنجیره ای زیادی ایجاد کنند که ممکن است باعث آسیب سلولی شود. خوشبختانه بدن انسان می تواند چنین مکانیسم های ایمنی ساخته شده را ایجاد کند، با این وجود هنگام نصب گونه های اکسیژن واکنشی، یا ROS قادر به خنثی شدن نیستند، یک شعله کوچک را که در هنگام تزریق با اکسیژن خارج می شود، پیش بینی می شود، .
برای ادامه دادن به استعاره شعله، محصول نهایی که توانایی خنثی کردن اثر ROS یا گونه های اکسیژن واکنشی را ندارد، آسیب و التهاب است؛ به عبارت دیگر، بدن انسان به معنای واقعی کلمه در آتش است. چیز فوق العاده آنتی اکسیدان هایی است که می تواند به شدت به مبارزه با این مسئله سلامتی کمک کند و این آنتی اکسیدان گلوتاتیون است. گرچه در 1889 یافت شده است، اثر آنتی اکسیدانی گلوتاتیون یکی از موضوعات جالب در مطالعات تحقیقاتی مدرن است.
استاد آنتی اکسیدان ها: گلوتاتیون
ماده قوی یک تری پپتید است که از سیستئین ، اسید گلوتامیک و گلیسین ایجاد می شود. گلوتاتیون به دلیل توانایی محافظت از بدن انسان در برابر ایجاد رادیکال های آزاد ، در نهایت می تواند به ارتقا promote سیستم ایمنی سالم کمک کند. بر اساس گزارش های علمی در 2015، مشخص شد که توانایی گلوتاتیون برای کارکردن همزمان با پراکسیدیدین و کاتالاز، باعث محافظت سلول در برابر پراکسید هیدروژن می شود. این فرمول ترکیبی با انواع اکسیژن واکنشی یا ROS عمل می کند. گلوتاتيون، پراكسيديديدين و كاتالاز عناصر ضروري در افزايش هوموتازي سلولي هستند كه فرآيند ضروري سلول ها، بافت ها و اندام هاي سالم است.
علاوه بر این، گلوتاتیون کلیه ساختار سیستم ایمنی بدن و عملکرد را با استفاده از اثر مهم آن بر عملکرد لنفوسیت ها افزایش می دهد. بر اساس گروه ایمونوشیمی، به درستی مکمل سطح گلوتاتیون در بدن انسان می تواند تا حد زیادی واکنش های ایمنی را افزایش دهد. به عنوان مثال، دو کارآزمايي تصادفي شده با کنترل دارونما نشان داد که درمان درماني بيماران مبتلا به ايمني با N-acetyl-cysteine يا NAC در هر دو مورد منجر به رشد قابل توجهي در بيشتر پروتئين هاي ايمونولوژيک شده بود که شامل جوانه زني کامل فعالیت سلولی قاتل طبیعی. N-acetyl-cysteine، یا NAC، از گوگرد از گلوتاتیون استفاده می کند و آن را با مولکول های سمی ترکیب می کند که سپس به محلول در آب تبدیل می شوند و در بدن انسان تخلیه می شوند.
گلوتاتیون همچنین توانایی احیاء اسید لیپوئیک و همچنین بازیافت ویتامین C و E را دارد که برای ایجاد فرآیندهای سیستم خاص از طریق ارسال الکترونها برای خنثی سازی رادیکال های آزاد ضروری است. بر اساس یک مطالعه تحقیق از مجله PLOS ONE، گلوتاتيون مبتلا به ديابت نوع دو، يا T2DM و ميکوباکتريوم توبرکلوزيس. به طور معمول افراد مبتلا به سیستم ایمنی ضعیف تمایل دارند که تماس بیشتری با M. tb یا mycobacterium tuberculosis، بیماری یا عفونت داشته باشند. علاوه بر این افراد مبتلا به دیابت نوع 2 یا T2DM دو تا سه برابر بیشتر نسبت به افراد بدون T2DM مبتلا به سل هستند. تحقیقات همچنین نشان داد که افزایش سطح گلوتاتیون در ماکروفاژها جدا شده از بیماران مبتلا به T2DM منجر به بهبود کنترل بیماری M.Tb یا عفونت شد. این نتایج نشان می دهد که سطح پایین گلوتاتیون در بیماران مبتلا به T2DM موجب افزایش احتمال ابتلا به بیماری M. tb یا عفونت می شود. علاوه بر این، وابسته به دیروز غزی در مدارس پزشکی برتون و ساسکساسترس اکسیداتیو در نهایت می تواند ساختاری و عملکرد سیستم ایمنی بدن ضعیف را به همراه داشته باشد.
خوشبختانه، گلوتاتیون نقش مهمی در تقویت و کنترل ایمنی دارد. به طور مثال، گلوتاتیون برای فرآیندهای ذاتی و انعطاف پذیر در سیستم ایمنی ضروری است، از جمله proliferation T-lymphocyte، فعالیت فاگوسیتیک نوتروفیل های چند هسته ای و سلول های دندریتیک، که می تواند اساسی باشد زیرا اینها از سلول های ارائه دهنده آنتی ژن تشکیل شده اند . ایمنی سلول مدیت شده شامل آنتی ژن های پروتئینی است که ابتدا در غشاهای اندوسیتیک ماکروفاژها و سلول های دندریتیک دژنراسیون ایجاد می کنند، بنابراین پپتیدهای کوچکتر روی سطح برای نشان دادن تکثیر سلول های اختصاصی آنتی ژن مشخص می شوند. علاوه بر این، گلوتاتیون باعث ایجاد سیتوکین ها می شود و لازم است که تولید اینترفرون گاما توسط سلول های دندریتیک حفظ شود، که برای حفاظت از پاتوژن های داخل سلولی از جمله میکوباکتری ها مهم است.
N-acetyl-cysteine یا NAC، که به لحاظ علمی به عنوان پیش ماده گلوتاتیون شناخته می شود، همچنین یک آنتی اکسیدان سلولی قوی است که به عنوان یک آنتی اکسیدان رسوب کننده رادیکال آزاد استفاده می شود. به طور معمول برای نقش آن در جلوگیری از سمیت استامینوفن، NAC ، یا -N-استیل-سیستئین ، نشان داده شده است که دارای چندین مزیت سلامتی و سلامتی است. مطابق با مجله سلولی، NAC به حمایت از یک پاسخ التهابی سالم کمک می کند و ممکن است به طور معناداری بر شرایط انسانی و زایمان زودرس موثر باشد. تحقیقات انجام شده نشان می دهد که در زنان مبتلا به زایمان زودرس و واژینوز باکتریایی، 0.6 گرم NAC در روز به صورت خوراکی همراه با پروژسترون پس از هفته 16 بارداری محافظت شده در برابر عود زودرس زودرس و بهبود نتیجه نوزادان. در نتیجه، اثرات مثبت NAC بر روی ساخت عضله نیز شناسایی شد. پس از سه دقیقه انقباضات پایدار، یک افزایش 15 درصد افزایش یافت و نشان داد که چگونه NAC نقش اساسی در بهبود ساختار عضلانی و کاهش خستگی کلی در طول کار دارد.
محققان همچنین کشف کردند که NAC یا استیل-سیستئین ممکن است به نفع کسانی باشد که به سندرم تخمدان پلی کیستیک یا PCOS مبتلا هستند. PCOS یا سندرم تخمدان پلی کیستیک ، یک بیماری شایع مربوط به غدد درون ریز است که تقریباً 5 تا 10 درصد از زنان در سن باروری را تحت تأثیر قرار می دهد. در چنین بیمارانی ، بیشتر خطر ابتلا به سندرم متابولیک وجود دارد ، جایی که استفاده از NAC به بازگرداندن سطح انسولین و حساسیت سالم کمک می کند.
دکتر الکس جیمنز Insight
به دلیل نقش اساسی آن در دستیابی و حفظ سلامتی و سلامتی کلی، گلوتاتیون به عنوان "استاد آنتی اکسیدان" شناخته می شود. در حالی که بدن انسان توان تولید گلوتاتیون خود را دارد، تغذیه نامناسب، آلودگی، سموم، مصرف بیش از حد از داروها و / یا داروها، استرس، تروما، پیری، بیماری و تابش می تواند سطح طبیعی گلوتاتیون را کاهش دهد. این امر به نوبه خود باعث می شود افرادی که بیشتر از آسیب به سلول از استرس اکسیداتیو، رادیکال های آزاد، عفونت ها و سرطان حساس هستند. مکمل Glutathione می تواند مزایای زیادی در بدن انسان داشته باشد. همراه با گزینه های درمان جایگزین، مانند مراقبت از کایروپراکتیک، سطح گلوتاتیون می تواند یک بار دیگر برای بهبود وضعیت سلامتی تنظیم شود.
علاوه بر این، متخصصان مراقبت های بهداشتی پیشنهاد کرده اند که استفاده از مکمل های گلوتاتيون همراه با سایر گزینه های درمان جایگزین، مانند مراقبت از کایروپراکتیک، برای بهبود بیشتر سلامت و سلامت کلی. آنتی اکسیدان ها در جهت حفظ حداکثر رفاه و جلوگیری از واکنش زنجیره ای رادیکال های آزاد که موجب آسیب یا آسیب به سلول می شوند، مهم هستند. آنتی اکسیدان های قدرتمند مانند گلوتاتیون، همانطور که قبلا در بالا ذکر شد، در نهایت کمک می کنند که تنظیم این رادیکال های آزاد را تنظیم کرده و پاسخ سیستم ایمنی سالمتری را ارائه دهند. مطالعات تحقیقاتی آن را یافته اند مراقبت از کایروپراکتیک همچنین ممکن است نقش مهمی در این فرآیند داشته باشد، به طور طبیعی باعث افزایش فعالیت آنتی اکسیدانها در بدن انسان می شود. مراقبت های کایروپراکتیک یک روش درمان ایمن و موثر است که از تنظیمات ستون فقرات و دستکاری دستی برای اصلاح ناهماهنگی های ستون فقرات یا سوکسوکساسیون استفاده می کند تا بدن انسان بدون استفاده از دارو / دارو و / یا مداخلات جراحی به طور طبیعی خود را درمان کند.
سرانجام ، آنتی اکسیدان ها ویژگی های بیولوژیکی خود را از طریق بسیاری از مزایای سلامتی نشان می دهند ، که ممکن است اکنون بیش از هر زمان دیگری با هجوم استرس ، بیماری و آلودگی در دنیای مدرن ما ، که همگی در آسیب سلول و / یا آسیب سلول موثر هستند ، لازم باشد. . گلوتاتیون و پیش ماده آن ، NAC ، یا NN-استیل-سیستئین ، همچنان قدرتمند خود را در حوزه آنتی اکسیدان ها نشان می دهند. همراه با گزینه های درمانی جایگزین ، مانند مراقبت های کایروپراکتیک ، افراد می توانند از تمام مزایایی که این آنتی اکسیدان قدرتمند ارائه می دهد استفاده کنند. دامنه اطلاعات ما محدود به عمل جراحی و همچنین آسیب های ستون فقرات و شرایط است. برای بحث در مورد موضوع ، لطفا در صورت تمایل از دکتر خیمنز بپرسید یا با ما تماس بگیرید915-850-0900.
دکتر الکس جیمنز سرپرستی می کند
موارد اضافی: درد پشت
درد پشت یکی از شایع ترین علل ناتوانی و از دست رفته روز در کار در سراسر جهان است. به عنوان حقيقت، درد پشتي به عنوان دومين شايعترين دلايل بازديد پزشکي، نسبت به عفونت فوقاني تنفسي کمتر است. تقریبا 80 درصد از جمعیت حداقل یک بار در طول زندگی خود نوعی کمر درد را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده ای است که از استخوان ها، مفاصل، لیگامان ها و ماهیچه ها در میان دیگر بافت های نرم تشکیل شده است. به همین دلیل، آسیب و / یا شرایط شدید، مانند دیسک های فتق دیسک، در نهایت می تواند منجر به علائم درد پشت شود. آسیب های ورزشی یا آسیب های ناشی از تصادفات خودرو اغلب اغلب علت درد پشت هستند، اما گاهی اوقات ساده ترین حرکات می توانند نتایج دردناکی داشته باشند. خوشبختانه، گزینه های درمان جایگزین، مانند مراقبت از کیهان پراکسی، می تواند به کاهش درد در استفاده از تنظیمات ستون فقرات و دستکاری دست کمک کند و در نهایت بهبود تسکین درد را کاهش دهد.
علم کیهان شناسی دکتر الکساندر جیمنز نگاهی به استرس اکسیداتیو، این چه چیزی است، چگونه بدن و دفاع آنتی اکسیدان را برای بهبود وضعیت بر بدن تاثیر می گذارد.
دکتر اسرار بیبرن، دکتر 1 Umit Murat Sahiner MD، 1 Cansin Sackesen MD، 1 Serpil Erzurum MD، 2 و Omer Kalayci، MD1
چکیده: گونه های اکسیژن واکنش پذیر (ROS) توسط ارگانیسم های زنده در نتیجه متابولیسم سلولی طبیعی و عوامل محیطی مانند آلاینده های هوا یا دود سیگار تولید می شوند. ROS مولکولهای بسیار واکنشی هستند و می توانند به ساختارهای سلولی مانند کربوهیدرات ها ، اسیدهای نوکلئیک ، لیپیدها و پروتئین ها آسیب برسانند و عملکرد آنها را تغییر دهند. تغییر در تعادل بین اکسیدان ها و آنتی اکسیدان ها به نفع اکسیدان ها ، استرس اکسیداتیو نامیده می شود. تنظیم وضعیت کاهش و اکسیداسیون (ردوکس) برای زنده ماندن سلول ، فعال سازی ، تکثیر و عملکرد اندام بسیار مهم است. ارگانیسم های هوازی سیستم های آنتی اکسیدانی یکپارچه ای دارند که شامل آنتی اکسیدان های آنزیمی و غیر آنزیمی است که معمولاً در جلوگیری از اثرات مضر ROS موثر هستند. با این حال ، در شرایط پاتولوژیک ، سیستم های آنتی اکسیدانی می توانند غرق شوند. استرس اکسیداتیو به بسیاری از بیماری ها و بیماری های پاتولوژیک ، از جمله سرطان ، اختلالات عصبی ، تصلب شرایین ، فشار خون بالا ، ایسکمی / پرفیوژن ، دیابت ، سندرم زجر تنفسی حاد ، فیبروز ریوی ایدیوپاتیک ، بیماری انسدادی مزمن ریوی و آسم کمک می کند. در این بررسی ، ما سیستم اکسیدان سلولی و آنتی اکسیدانی را خلاصه می کنیم و در مورد اثرات سلولی و مکانیسم های استرس اکسیداتیو بحث می کنیم.
گونه های اکسیژن واکنش پذیر (ROS) توسط ارگانیسم های زنده و در نتیجه متابولیسم سلولی طبیعی تولید می شوند. در غلظت های کم تا متوسط ، آنها در فرآیندهای سلول های فیزیولوژیکی عمل می کنند ، اما در غلظت های بالا ، تغییرات نامطلوبی در اجزای سلول مانند لیپیدها ، پروتئین ها و DNA ایجاد می کنند. 1-6 تغییر تعادل بین اکسیدان / آنتی اکسیدان به نفع اکسیدان استرس اکسیداتیو نامیده می شود. استرس اکسیداتیو به بسیاری از بیماری های پاتولوژیک کمک می کند ، از جمله سرطان ، اختلالات عصبی ، آترواسکلروز 7-10 ، فشار خون بالا ، ایسکمی / پرفیوژن ، دیابت 11-14 ، سندرم دیسترس تنفسی حاد ، فیبروز ریوی ایدیوپاتیک ، بیماری انسدادی مزمن ریوی ارگانیسم های هوازی دارای سیستم های آنتی اکسیدانی یکپارچه هستند ، که شامل آنتی اکسیدان های آنزیمی و غیر آنزیمی است که معمولاً در جلوگیری از اثرات مضر ROS موثر هستند. با این حال ، در شرایط پاتولوژیک ، سیستم های آنتی اکسیدانی می توانند غرق شوند. در این بررسی ، ما به طور خلاصه سیستم های اکسیدان و آنتی اکسیدان های سلولی و تنظیم وضعیت کاهش و اکسید کننده (ردوکس) در حالت های بهداشتی و بیماری ها را بیان می کنیم.
اکسیدان ها
منابع اندوژن ROS
ROS از اکسیژن مولکولی به عنوان یک نتیجه از متابولیسم طبیعی سلول تولید می شود. ROS را می توان به گروه های 2 تقسیم کرد: رادیکال های آزاد و غیر رادیکال ها. مولکول هایی که حاوی یک یا چند الکترون ناپیوسته هستند و به این ترتیب واکنش پذیری به مولکول می دهند، رادیکال های آزاد نامیده می شوند. هنگامی که رادیکال های آزاد 2 الکترونهای ناسازگار خود را به اشتراک می گذارند، اشکال غیر رادیکال ایجاد می شود. 3 عمده ROS که اهمیت فیزیولوژیکی دارند عبارتند از سوپراکسید آنیون (O22.)، هیدروکسیل رادیکال (OH) و پراکسید هیدروژن (H2O2). ROS در جدول 1 خلاصه شده است.
آنیون سوپراکسید با افزودن 1 الکترون به اکسیژن مولکولی تشکیل می شود .22 این فرآیند توسط نیکوتین آدنین دی نوکلئوتید فسفات [NAD (P) H] اکسیداز یا اکسیداز گزانتین یا با سیستم انتقال الکترون میتوکندری ایجاد می شود. محل اصلی تولید آنیون سوپراکسید میتوکندری است ، ماشین آلات سلول برای تولید آدنوزین تری فسفات. به طور معمول ، الکترون ها از طریق زنجیره انتقال الکترون میتوکندری برای کاهش اکسیژن به آب منتقل می شوند ، اما تقریباً 1 تا 3٪ از کل الکترون ها از سیستم نشت می کنند و سوپراکسید تولید می کنند. NAD (P) H اکسیداز در لکوسیتهای پلی مورفونوکلئر ، مونوسیتها و ماکروفاژها یافت می شود. پس از فاگوسیتوز ، این سلول ها یک انفجار سوپراکسید تولید می کنند که منجر به فعالیت ضد باکتری می شود. سوپراکسید با عملکرد سوپراکسید دیسموتازها به پراکسید هیدروژن تبدیل می شود (SODs ، EC 1.15.1.1). پراکسید هیدروژن به راحتی در غشا plas پلاسما پخش می شود. پراکسید هیدروژن نیز توسط گزانتین اکسیداز ، اسید آمینه اکسیداز و NAD (P) H oxidase 23,24،2 و در پراکسیزوم ها با مصرف اکسیژن مولکولی در واکنش های متابولیکی تولید می شود. در پی واکنشهایی به نام واکنشهای Haber Weiss و Fenton ، H2O2 می تواند در حضور فلزات انتقال دهنده مانند Fe21 یا Cu21.25 به OHXNUMX تجزیه شود.
Fe31 + .O2 ؟ Fe2 + O2 هابر ویس
Fe2 + H2O2 ؟ Fe3 + OH + .OH واکنش فنتون
O2 خود نیز می تواند با H2 O2 واکنش نشان دهد و باعث تولید OH26,27،XNUMX رادیکال هیدروکسیل واکنش پذیرترین ماده ROS است و می تواند به پروتئین ها ، چربی ها و کربوهیدرات ها و DNA آسیب برساند. همچنین می تواند با گرفتن الکترون از اسیدهای چرب اشباع نشده اشباع شده ، پراکسیداسیون لیپیدها را شروع کند.
آنزیم های گرانولوسیتی بیشتر واکنش H2O2 را از طریق ائوزینوفیل پراکسیداز و میلوپراکسیداز (MPO) گسترش می دهند. در نوتروفیل های فعال شده ، H2O2 توسط MPO مصرف می شود. در حضور یون کلرید ، H2O2 به اسید هیپوکلروس (HOCl) تبدیل می شود. HOCl بسیار اکسیداتیو است و نقش مهمی در از بین بردن عوامل بیماری زا در مجاری تنفسی دارد .28 با این حال ، HOCl همچنین می تواند با DNA واکنش داده و فعل و انفعالات DNA پروتئین ایجاد کند و محصولات اکسیداسیون پیریمیدین تولید کند و به پایه های DNA کلراید اضافه کند. 29,30،31 ائوزینوفیل پراکسیداز و MPO همچنین با اصلاح پروتئین ها توسط هالوژناسیون ، نیتراسیون و پیوندهای متقابل پروتئین از طریق رادیکال های تیروزیل به استرس اکسیداتیو کمک می کنند. 33-XNUMX
دیگر رادیکال های آزاد شده از اکسیژن، رادیکال های پراکسیل (ROO $) هستند. ساده ترین شکل این رادیکال ها رادیکال هیدروپروکسیل (HOO $) است و در پراکسیداسیون اسید های چرب نقش دارد. رادیکال های آزاد می توانند واکنش های زنجیره ای پراکسیداسیون لیپید را با انتزاع یک اتم هیدروژن از یک کربن متیلن جانبی پیوند ایجاد کنند. سپس رادیکال چربی با اکسیژن واکنش میدهد تا رادیکال پراکسیل را تولید کند. رادیکال پراکسیل یک واکنش زنجیره ای را آغاز می کند و اسیدهای چرب اشباع نشده را به هیدروپراکسید های لیپیدی تبدیل می کند. هیدروپراکسید لیپید بسیار ناپایدار است و به محصولات ثانویه مانند آلدئیدها (مانند 4-hydroxy-2,3-nonenal) و مالون دی آلدئید ها (MDAs) تجزیه می شود. ايزوپروستانها گروهي ديگر از محصولات پراكسيداسيون چربي هستند كه از طريق پراكسيداسيون اسيد آراكيدونيك توليد مي شوند و همچنين در پلاسما و غلظت هاي تنفس آسم نيز افزايش يافته است. اكسيداسيون ليپيدها باعث كاهش غلظت غشاء سلولي و منجر به بازسازي ساختار غشايي مي شود. .
پراکسید هیدروژن، رادیکال سوپراکسید، گلوتاتیون اکسید شده (GSSG)، MDA، ایزوپروستان، کربونیل و نیتروتیروسین را می توان به راحتی از نمونه های پلاسما، خون و یا لارو برونکوه آلوئولار به عنوان بیومارکرهای اکسیداسیون با روش های استاندارد اندازه گیری کرد.
منبع خارج اکسیدان
دود سیگار
دود سیگار شامل بسیاری از اکسیدان ها و رادیکال های آزاد و ترکیبات ارگانیک مانند سوپراکسید و اکسید نیتریک است. 36 علاوه بر این، استنشاق سیگار سیگار در ریه همچنین برخی از مکانیسم های درونی مانند تجمع نوتروفیل ها و ماکروفاژها را فعال می کند که بیشتر آسیب اکسیدان را افزایش می دهد .
قرار گرفتن در معرض ازن
قرار گرفتن در معرض ازن می تواند باعث پراکسیداسیون لیپید شود و موجب نفوذ نوتروفیل ها به اپیتلیوم هوایی شود. در معرض قرار گرفتن در معرض کوتاه مدت قرار گرفتن در معرض ازن همچنین باعث آزاد شدن واسطه های التهابی مانند پروتئین MPO، پروتئین های کاتیونی ائوزینوفیل و همچنین لکتات دهیدروژناز و آلبومین می شود. 37 حتی در افراد سالم، قرار گرفتن در معرض ازن باعث کاهش عملکرد توده های ریوی می شود. 38 Cho et al39 نشان داده است ذرات جامد (مخلوطی از ذرات جامد و قطره های مایع که در هوا معلق هستند) کاهش اکسیژن را کاهش می دهد.
هیپوکسیا
هیپوکسیا به شرایط سطوح اکسیژن بالاتر نسبت به عادی فشار جزئی اکسیژن در ریه ها و دیگر بافت های بدن اشاره دارد. این منجر به تولید بیشتر از گونه های واکنش پذیر اکسیژن و نیتروژن می شود. 40,41
تابش یونیزه
تابش یونیزه کننده ، در حضور O2 ، رادیکال های هیدروکسیل ، سوپراکسید و رادیکال های آلی را به پراکسید هیدروژن و هیدروپراکسیدهای آلی تبدیل می کند. این گونه های هیدروپراکسید از طریق واکنشهای فنتون با یونهای فلزی فعال اکسیداسیون ، مانند Fe و Cu واکنش نشان می دهند و بنابراین استرس اکسیداتیو ایجاد می کنند. 42,43،44 Narayanan و همکاران 2 نشان دادند که فیبروبلاست هایی که در معرض ذرات آلفا قرار داشتند ، افزایش قابل توجهی در O2 2 و H2O44 داخل سلول داشتند تولید از طریق NADPH اکسیداز متصل به غشا پلاسما .1 مولکول های انتقال سیگنال ، مانند کیناز 2 و 1 با تنظیم سیگنال خارج سلول (ERK2 / 38) ، کیناز ترمینال N-j-c (JNK) و p1 ، و عوامل رونویسی ، مانند پروتئین فعال کننده 1 (AP-53) ، فاکتور هسته ای kB (NF-kB) و p45 فعال می شوند ، که منجر به بیان ژن های مربوط به واکنش تابش می شود. فوتون های 50-8 اشعه ماوراlet بنفش A (UVA) واکنش های اکسیداتیو ایجاد می کنند با تحریک حساسیت به نور درون زا ، مانند پورفیرین ها ، NADPH اکسیداز و ریبوفلاوین ها. 7,8-Oxo-8،1- dihydroguanine (51-oxoGua) اصلی ترین محصول اکسیداسیون DNA با واسطه UVA است که در اثر اکسیداسیون رادیکال OH ، اکسیدان های 52,53 الکترون و اکسیژن منفرد ایجاد می شود که عمدتا با گوانین واکنش نشان می دهد. 54 تشکیل گوانین کاتیون رادیکال در DNA جدا شده نشان داده شده است که به طور م throughثر از طریق تأثیر مستقیم تابش یونیزان رخ می دهد. XNUMX/XNUMX پس از قرار گرفتن در معرض تابش یونیزان ، سطح داخل سلولی گلوتاتیون (GSH) برای مدت کوتاه کاهش می یابد اما دوباره افزایش می یابد. XNUMX
یون سنگین فلز
یونهای سنگین فلزات مانند آهن، مس، کادمیوم، جیوه، نیکل، سرب و آرسنیک می توانند باعث ایجاد نسل رادیکال های واکنشی و ایجاد آسیب سلولی از طریق کاهش فعالیت های آنزیم از طریق پراکسیداسیون لیپید و واکنش با پروتئین های هسته ای و DNA.55
یکی از مهمترین مکانیزمهای تولید رادیکالهای آزاد با واسطه فلز ، از طریق واکنش نوع Fenton است. یون سوپراکسید و پراکسید هیدروژن می توانند با فلزات انتقالی مانند آهن و مس از طریق واکنش فلز Haber Weiss / Fenton کاتالیز شده و رادیکال های OH را ایجاد کنند.
علاوه بر مکانیسم های نوع Fenton و Haber Weiss ، یون های فلزی خاصی می توانند با مولکول های سلولی به طور مستقیم واکنش نشان دهند و رادیکال های آزاد مانند رادیکال های تیول تولید کنند یا مسیرهای سیگنالینگ سلول را القا کنند. این رادیکال ها ممکن است با مولکول های دیگر تیول نیز واکنش نشان دهند و O22 تولید کنند. به H22O2 تبدیل می شود که باعث ایجاد رادیکال اکسیژن اضافی می شود. برخی از فلزات ، مانند آرسنیت ، با فعال سازی سیستم های تولید رادیکال در سلول ها ، باعث ایجاد ROS به طور غیرمستقیم می شوند
آرسنیک یک عنصر بسیار سمی است که انواع ROS را تولید می کند ، از جمله سوپراکسید (O2 2) ، اکسیژن واحد (1O2) ، رادیکال پراکسیل (ROO) ، اکسید نیتریک (NO) ، پراکسید هیدروژن (H2O2) و رادیکال های پراکسیل دی متیلارزیک [( CH3) 2AsOO] .57-59 ترکیبات آرسنیک (III) می توانند از طریق اتصال آنزیم های آنتی اکسیدان ، به ویژه آنزیم های وابسته به GSH ، مانند گلوتاتیون-ترانسفرازها (GST) ، گلوتاتیون پراکسیداز (GSH-Px) و GSH ردوکتاز را مهار کنند. - به گروه های سولفیدریل ( SH) آنها. 60,61،XNUMX
سرب باعث افزایش پراکسیداسیون لیپید می شود. 62 کاهش قابل توجهی در فعالیت SOD بافت و مغز GPx پس از قرار گرفتن در معرض سرب گزارش شده است. 63,64 جایگزینی روی، که به عنوان کافاکور برای بسیاری از آنزیم ها توسط سرب عمل می کند، منجر به غیر فعال شدن چنین آنزیم ها می شود. قرار گرفتن در معرض سرب می تواند مهار GST را با تأثیر تيول های بافت ايجاد کند.
ROS تولید شده توسط واکنش های کاتالیزوری فلز می تواند پایگاه های DNA را تغییر دهد. سه جایگزین پایه، G / C، G / T و C / T می تواند به علت آسیب اکسیداتیو توسط یون های فلزی مثل Fe21، Cu21 و Ni21 رخ دهد. Reid et al65 نشان داد که G / C به طور عمده توسط Fe21 تولید می شد در حالیکه C / T جایگزینی توسط Cu21 و Ni21 بود.
آنتی اکسیدان ها
بدن انسان با انواع آنتی اکسیدان ها مجهز شده است که برای مقابله با اثر اکسیدان ها مناسب است. برای تمام اهداف عملی، این را می توان به دسته های 2 دسته بندی: آنزیمی (جدول 2) و nonenzymatic (جدول 3).
آنتی اکسیدان آنزیمی
آنتی اکسیدان های آنزیمی عمده ریه ها عبارتند از SODs (EC 1.15.1.11)، کاتالاز (EC 1.11.1.6) و GSH-Px (EC 1.11.1.9). علاوه بر این آنزیم های مهم دیگر آنتی اکسیدان ها، از جمله همای اکسیژناز 1 (EC 1.14.99.3) و پروتئین های بازسازی مانند تیرودوکسین ها (TRXs، EC 1.8.4.10)، پراکسی اوردوکسین ها (PRXs، EC 1.11.1.15) و گلوتاراکسین ها نیز یافت شده است نقش مهمی در دفاع از آنتی اکسیدان ریه بازی کند.
از آنجا که سوپراکسید اولیه ROS تولید شده از منابع مختلف است، تقسیم آن توسط SOD اهمیت اولیه برای هر سلول است. تمام اشکال 3 SOD، یعنی CuZn-SOD، Mn-SOD، و EC-SOD، به طور گسترده ای در ریه انسان بیان می شود. Mn-SOD در ماتریکس میتوکندری موضعی قرار دارد. EC-SOD عمدتا در ماتریکس خارج سلولی قرار دارد، به خصوص در مناطقی که مقادیر بالایی از فیبرهای کلاژن نوع I و در اطراف عروق ریه و سیستمیک دارند. همچنین در اپیتلیوم برونش، اپیتلیوم آلوئولار و ماکروفاژهای آلوئولار تشخیص داده شده است. 66,67 به طور کلی، CuZn-SOD و Mn-SOD به طور کلی تصور می شود که عملکردهای فله ای رادیکال های سوپراکسید عمل کنند. سطح نسبتا بالا EC-SOD در ریه با اتصال خاص خود به اجزای ماتریکس خارج سلولی ممکن است یک جزء اساسی حفاظت از ماتریکس ریه را نمایان سازد. 68
H2O2 که توسط عمل SOD ها تولید می شود و یا عمل اکسیداز ها مانند اکسیداز زانتین به کاتالاز و GSH-Px به آب کاهش می یابد. کاتالاز به عنوان یک tetramer از مونومرهای یکسان 4 وجود دارد که هر کدام شامل گروه هام در محل فعال می باشد. تجزیه H2O2 از طریق تبدیل از ترکیبات 2 از کاتالاز-آهنریاتالاز (آهن هماهنگ شده به آب) و ترکیب I (آهن متشکل از یک اتم اکسیژن) انجام می شود. کاتالاز همچنین NADPH را به عنوان یک معادل کننده کاهش می دهد تا از مهار اکسیداتیو آنزیم (تشکیل ترکیب دوم) توسط H2O2 جلوگیری کند زیرا آن را به آب کاهش می دهد. 69
آنزیم هایی که در چرخه رادیواکتیو برای کاهش H2O2 و هیدروپراکسید لیپیدها تولید می شوند (تولید شده در نتیجه پراکسیداسیون لیپید غشاء) شامل GSH-Pxs.70 GSH-Pxs یک خانواده از آنزیم های tetrameric است که حاوی selenocysteine منحصر به فرد آمینو اسید درون سایت های فعال و استفاده از تیول های کم وزن مولکولی مانند GSH، برای کاهش H2O2 و پراکسید های چربی به الکل های مربوطه آنها. چهار GSH-Pxs توصیف شده، کدگذاری شده توسط ژن های مختلف: GSH-PX-1 (سلول GSH-Px) در همه جا قرار دارد و H2O2 و پراکسید اسید چرب را کاهش می دهد، اما لیپید های پراکسل نخونده نشده است. 71 لیپید های استرین شده توسط GSH غشایی -Px-4 (هیدروپراکسید فسفولیپید GSH-Px)، که می تواند از چندین تیئول با وزن مولکولی کمتر به عنوان کاهش معادل استفاده کند. GSH-Px-2 (GSH-Px دستگاه گوارش) در سلولهای اپیتلیال دستگاه گوارش قرار دارد که در آن برای کاهش پراکسید های غذایی استفاده می شود. 72 GSH-Px-3 (GSH-Px خارج سلولی) تنها عضو خانواده GSH-Px است که در محفظه خارج سلولی و به نظر می رسد یکی از مهم ترین آنزیم آنتی اکسیدان خارج سلولی در پستانداران است. از این موارد، GSH-Px خارج سلولی به طور گسترده ای در ریه انسان بررسی می شود. 73
علاوه بر این، دفع H2O2 با چندین آنزیم حاوی تيول، یعنی TRXs (TRX1 و TRX2)، تيورودوکسين ردوکتاز (EC 1.8.1.9) (TRRs)، PRX ها (که داراي تيورودوکسين پراکسيداز) و گلوتاراکسوين ها هستند. 74
دو TRXs و TRRs در سلول های بنیادی موجود در هر دو سیتوزول و میتوکندری وجود دارد. در ریه، TRX و TRR در اپیتلیوم برونش و الوئول و ماکروفاژها بیان می شود. شش PRX مختلف در سلول های انسانی یافت شده اند، که در بخش بندی های فراساختاری آنها متفاوت است. مطالعات تجربی نشان می دهد اهمیت PRX VI در حفاظت از اپیتلیوم آلوئولار. ریه انسانی تمام PRX ها را در اپیتلیوم برونش، اپیتلیوم آلوئولی و ماکروفاژها بیان می کند. اخیرا 75 PRX V به عنوان یک ردوکتاز پراکسینیتریت، 76 عمل می کند که بدین معنی است که می تواند به عنوان یک ترکیب محافظتی بالقوه در ایجاد آسیب ریه، .77
به این آنتی اکسیدان ها رایج است که نیازمند NADPH به عنوان معادل کاهش است. NADPH کاتالاز را در فرم فعال حفظ می کند و به عنوان کافاکور توسط TRX و GSH ردوکتاز (EC 1.6.4.2) استفاده می شود که GSSG را به GSH تبدیل می کند که یک سوبسترا برای GSH-Pxs است. Intracellular NADPH، به نوبه خود، توسط کاهش NADP1 توسط گلوکز-6-فسفات دهيدروژناز، اولين و آنزيم محدود کننده سرعت پروتئين فسفات پتاسيم، در طي تبديل گلوکز-6-فسفات به 6-فسفوگلاوکنولاکتون توليد مي شود. با تولید NADPH، گلوکز-6-فسفات دهیدروژناز یک تعیین کننده حیاتی از ظرفیت بافر آمیلوئید سیتواستولی (GSH / GSSG) می باشد و بنابراین می تواند یک آنزیم آنتی اکسیدانی ضروری و پایدار باشد. 78,79
GSTs (EC 2.5.1.18) ، خانواده آنزیم های دیگر آنتی اکسیدان ، متابولیت های ثانویه مانند آلدئیدهای غیر اشباع ، اپوکسیدها و هیدروپراکسیدها را غیرفعال می کند. سه خانواده اصلی GST توصیف شده است: GST سیتوزولی ، GST میتوکندریایی ، 80,81،82 و GST میکروزومی وابسته به غشا که در متابولیسم ایکوزانوئید و GSH نقش دارد. 83 هفت کلاس GST سیتوزولی در پستانداران مشخص می شود ، Alpha ، Mu ، Pi ، Sigma ، Theta ، Omega و Zeta.86-1 در شرایط غیر استرس ، GST های کلاس Mu و Pi به ترتیب با کینازهای Ask87 و JNK تعامل دارند و این کینازها را مهار می کنند. 89-1 نشان داده شده است که GSTP89 از JNK در پاسخ به استرس اکسیداتیو. 1 GSTP90 همچنین از نظر جسمی با PRX VI تعامل دارد و منجر به بهبود فعالیت آنزیم PRX از طریق گلوتاتیونیلاسیون پروتئین اکسید شده می شود. XNUMX
آنتی اکسیدان های غیر انزمی
آنتی اکسیدان های غیرانسمیتی شامل ترکیبات کم مولکولی مانند ویتامین ها (ویتامین های C و E)، B-کاروتن، اسید اوریک و GSH، Tripeptide (Lg-glutamyl-L-cysteinyl-L-glycine) که حاوی تيول سولفیدریل) گروه.
ویتامین C (اسید اسکوربیک)
ویتامین C محلول در آب (اسید اسکوربیک) ظرفیت آنتی اکسیدانی فاز آب داخل سلولی و خارج سلولی را در ابتدا توسط رادیکال های آزاد اکسیژن آزاد می کند. این ویتامین E رادیکال های آزاد را به ویتامین E تبدیل می کند. سطح پلاسمای آن با سنی کاهش یافته است. 91,92
ویتامین E (a-توکوفرول)
ویتامین E محلول در چربی در داخل محل هیدروفوب غشای سلولی متمرکز است و اصلی ترین دفاعی در برابر آسیب غشای ناشی از اکسیدان است. ویتامین E الکترون را به رادیکال پراکسیل هدایت می کند که در طول پراکسیداسیون لیپید تولید می شود. a-توکوفرول فعال ترین شکل ویتامین E و آنتی اکسیدان مهار غشاء در سلول است. ویتامین E سبب آپوپتوز سلول های سرطانی و مهار تشکیلات رادیکال آزاد می شود. 93
گلوتاتیون
GSH بسیار فراوان است در تمام بخش های سلولی و مهمترین آنتی اکسیدان محلول است. نسبت GSH / GSSG یکی از عوامل اصلی استرس اکسیداتیو است. GSH اثرات آنتی اکسیدانی آن را به روش های مختلفی نشان می دهد. 94 با استفاده از عمل GSH-Px، پراکسید هیدروژن و لیپید پراکسید را سمیت می کند. GSH الکترون خود را به H2O2 اهدا می کند تا آن را به H2O و O2 کاهش دهد. GSSG دوباره به GSH توسط GSH reductase کاهش می یابد که از NAD (P) H به عنوان اهدا کننده الکترون استفاده می کند. GSH-Pxs همچنین برای حفاظت از غشای سلولی از پراکسیداسیون لیپید مهم است. کاهش گلوتاتیون پروتون ها را به ليپيدهای غشایی اهدا می کند و آنها را از حملات اکسيدان محافظت می کند. 95
GSH یک فاکتور برای چندین آنزیم های ضد سم زدایی مانند GSH-Px و ترانسفراز است. این نقش در تبدیل ویتامین C و E به اشکال فعال آنها دارد. GSH سلول ها را در برابر آپوپتوز محافظت می کند با تعامل با مسیرهای سیگنالینگ پرواپوپتوزی و آناپاپتوکتیک. 94 همچنین تعدادی از عوامل رونویسی مانند AP-1، NF-kB و Sp-1 را تنظیم و فعال می کند.
کاروتنوئیدها (ب کاروتن)
کاروتنوئیدها رنگدانه های موجود در گیاهان هستند. در ابتدا بکاروتن با واکنش های پراکسیل (ROO)، هیدروکسیل (OH) و سدوکسی (O22.) رادیکال ها واکنش نشان می دهد. 96 کاروتنوئیدها اثرات آنتی اکسیدانی خود را در فشار جزئی کم اکسیژن نشان می دهند اما ممکن است اثرات پروکسید کننده در اکسیژن بالاتر داشته باشند 97 هر دو کاروتنوئیدها و اسیدهای رتینوئیک (RA) قادر به تنظیم عوامل رونویسی می باشند. 98 ب کاروتن مهار فعالیت فعال NF-kB ناشی از اکسیدان و X-NULX و اینترلوکین (IL) -6 و فاکتور تولید ناباروری تومور است. کاروتنوئید ها نیز بر آپوپتوز سلول ها تاثیر می گذارند. اثرات ضد انعقادی RA در مطالعات مختلف نشان داده شده است. این اثر RA عمدتا توسط گیرنده های اسید رتینوئیک محسوب می شود و در میان انواع سلول ها متفاوت است. در سلولهای کارسینوماى ماموتى، گیرنده اسید رتینوئیک منجر به مهار رشد توسط القای توقف چرخه سلولی، آپوپتوز یا هر دو شد. 99,100
تأثیر استرس اکسیداتیو: مکانیسم های ژنتیکی ، فیزیولوژیکی و بیوشیمیایی
استرس اکسیداتیو زمانی اتفاق می افتد که تعادل بین آنتی اکسیدان ها و ROS به علت تخلیه آنتی اکسیدان ها یا انباشت ROS اختلال ایجاد شود. هنگامی که استرس اکسیداتیو رخ می دهد، سلول های تلاش برای مقابله با اثرات اکسیدان و بازگرداندن تعادل ردوکس با فعال یا خاموش کردن ژن کد آنزیم دفاعی، عوامل scription tran- و نسبت proteins.101,102 ساختاری بین گلوتاتیون اکسید شده و کاهش می یابد (2GSH / GSSG) یکی است از عوامل مهم استرس اکسیداتیو در بدن. تولید بیشتر ROS در بدن ممکن است ساختار DNA را تغییر دهد، منجر به تغییر پروتئین ها و چربی ها، فعال شدن چندین رونویسی فاکتور ناشی از استرس و تولید سیتوکین های ضد التهابی و ضد التهابی شود.
اثرات استرس اکسیداتیو بر DNA
ROS می تواند به چندین روش منجر به تغییرات DNA شود که شامل تخریب پایگاه ها، انشعاب های DNA یا دو رشته ای، پورین، پیریمیدین یا تغییرات قند، اصلاح، جهش، حذف یا انتقال، و پیوند متقابل با پروتئین ها است. بسیاری از این تغییرات DNA (شکل 1) به شدت مربوط به سرطان زایی، پیری و بیماری های نورودنژنتیک، قلبی عروقی و خود ایمنی هستند. دود تنباکو، فلزات ردوکس و فلزات غیر خوراکی مانند آهن، کادمیوم، کروم و آرسنیک نیز در ایجاد سرطان و پیری با ایجاد رادیکال های آزاد یا اتصال با گروه های تیئول دخیل هستند. تشکیل 8-OH-G آسیب شناخته شده ترین DNA است که از طریق استرس اکسیداتیو اتفاق می افتد و یک نشانگر بالقوه برای سرطان زایی است.
مناطق پروموتر ژن ها توالی اجماعی برای عوامل رونویسی دارند. این سایت های اتصال دهنده فاکتور رونویسی دارای توالی غنی از GC هستند که برای حملات اکسیدان حساس هستند. تشکیل DNA 8-OH-G در سایت های اتصال فاکتور رونویسی می تواند اتصال فاکتورهای رونویسی را تغییر داده و در نتیجه بیان ژن های مرتبط را تغییر دهد همانطور که برای توالی های هدف AP-1 و Sp-1 نشان داده شده است .103 علاوه بر 8-OH-G ، همچنین نشان داده شده است که 8,59،29-سیکلو -104 -دئوکسیادنوزین (سیکلو-دی) می تواند مانع رونویسی از ژن گزارشگر در سیستم سلولی شود اگر در یک جعبه TATA قرار داشته باشد. XNUMX پروتئین متصل به TATA با تغییر در خم شدن DNA رونویسی را آغاز می کند . اتصال پروتئین متصل به TATA ممکن است با وجود سیکلو دی اختلال ایجاد کند.
استرس اکسید کننده باعث بی ثباتی ریزماهواره می شود (تکرار تاندوم کوتاه). یونهای فعال فلز Redox، رادیکالهای هیدروکسیل باعث افزایش بیثباتی میکرو رله می شوند. 105 اگر چه شکست DNA های تک رشته ای ناشی از آسیب اکسیدان به راحتی توسط سلول ها قابل تحمل می شود، اختلالات DNA رشته ای ناشی از اشعه یونیزاسیون می تواند تهدید مهمی برای بقای سلول باشد. 106
Methylation در جزایر CpG در DNA یک مکانیزم مهم اپی ژنتیک است که ممکن است باعث خاموش شدن ژن شود. اکسیداسیون 5-MeCyt به 5-هیدروکسی متیل اوراسیل (5-OHMeUra) می توانید از طریق واکنش های دآمیناسیون / اکسیداسیون تیمین یا 5-هیدروکسی متیل سیتوزین intermediates.107 علاوه بر بیان ژن تعدیل رخ می دهد، متیلاسیون DNA نیز به نظر می رسد را تحت تاثیر قرار organization.108 کروماتین الگوهای متیلاسیون DNA ناشی از حملات اکسیداتیو همچنین بر فعالیت های تعمیر DNA تاثیر می گذارد.
اثرات استرس اکسیداتیو بر روی لیپید
ROS می تواند باعث پراکسیداسیون لیپیدها شود و آرایش دو لایه لیپیدی غشایی را مختل کند که ممکن است گیرنده ها و آنزیم های متصل به غشا را غیرفعال کرده و نفوذپذیری بافت را افزایش دهد. -109 110-Hydroxy-112-nonenal باعث کاهش GSH داخل سلولی و ایجاد پروکسید می شود ، 4،2 گیرنده فاکتور رشد اپیدرم را فعال می کند ، 113,114 و تولید فیبرونکتین را القا می کند .115 ، به عنوان نشانگرهای زیستی غیرمستقیم استرس اکسیداتیو استفاده شده است ، و افزایش سطح در میعانات تنفسی بازدم یا مایع شستشوی برونکوآلوئولار یا ریه بیماران انسداد مزمن ریوی یا افراد سیگاری نشان داده شده است. 116 117
اثرات استرس اکسیداتیو بر پروتئین
ROS می تواند باعث تکه تکه شدن زنجیره پپتید ، تغییر بار الکتریکی پروتئین ها ، اتصال متقابل پروتئین ها و اکسیداسیون اسیدهای آمینه خاص شود و بنابراین منجر به افزایش حساسیت به پروتئولیز توسط تخریب توسط پروتئازهای خاص می شود. 120 باقی مانده سیستئین و متیونین در پروتئین ها اکسیداسیون گروه های سولفیدریل یا باقیمانده های متیونین پروتئین ها باعث تغییرات ساختاری ، باز شدن پروتئین و تخریب می شود. 121-8,121 آنزیم هایی که دارای فلزات در محل های فعال یا نزدیک به آنها هستند نسبت به اکسیداسیون کاتالیزور فلز حساس ترند. نشان داده شده است که اصلاح اکسیداتیو آنزیم ها از فعالیت آنها جلوگیری می کند .123،124,125
در برخی موارد، اکسیداسیون خاصی از پروتئین ها ممکن است رخ دهد. به عنوان مثال، متیونین می تواند اکسید متیونین sulfoxide126 و فنیل آلانین به o-tyrosine127؛ گروه های سولفیدریل می توانند به منظور تشکیل اوره های دی سولفید اکسید شوند؛ گروه های 128 و کربونیل ممکن است به زنجیره جانبی پروتئین وارد شوند. اشعه گاما، اکسیداسیون کاتالیزوری فلز، HOCl، و ازن می تواند باعث تشکیل گروه کربونیل شود. 129
اثرات استرس اکسیداتیو بر انتقال سیگنال
ROS می تواند بیانگر چندین ژن درگیر در انتقال سیگنال باشد .1,130 نسبت زیاد برای GSH / GSSG برای محافظت از سلول در برابر آسیب اکسیداتیو مهم است. اختلال در این نسبت باعث فعال شدن فاکتورهای رونویسی حساس به اکسیداسیون ، مانند NF-kB ، AP-1 ، فاکتور هسته ای سلول های T فعال شده و فاکتور 1 القا hyp کننده هایپوکسی می شود که در پاسخ التهابی نقش دارند. فعال شدن فاکتورهای رونویسی از طریق ROS با آبشارهای انتقال سیگنال که اطلاعات را از خارج به داخل سلول منتقل می کنند ، حاصل می شود. گیرنده های تیروزین کیناز ، بیشتر گیرنده های فاکتور رشد ، مانند گیرنده فاکتور رشد اپیدرمی ، گیرنده فاکتور رشد اندوتلیال عروقی و گیرنده فاکتور رشد مشتق از پلاکت ، پروتئین تیروزین فسفاتازها و کینازهای سرین / ترئونین از اهداف ROS هستند .131-133 کینازهای تنظیم شده با سیگنال خارج سلولی ، JNK و p38 ، که اعضای خانواده پروتئین کیناز فعال شده با میتوژن هستند و در چندین فرآیند از جمله تکثیر ، تمایز و آپوپتوز در سلول دخیل هستند ، همچنین توسط اکسیدان ها قابل تنظیم هستند.
تحت شرایط استرس اکسیداتیو ، بقایای سیستئین در محل اتصال DNA c-Jun ، برخی از زیرواحدهای AP-1 ، و kB کیناز مهاری تحت S-glutathiolation برگشت پذیر قرار می گیرند. گزارش شده است که گلوتاردوکسین و TRX نقش مهمی در تنظیم مسیرهای سیگنالینگ حساس به ردوکس دارند ، مانند NF-kB و AP-1 ، پروتئین کیناز فعال شده با میتوژن p38 و JNK.134 137
NF-kB می تواند در پاسخ به شرایط استرس اکسیداتیو ، مانند ROS ، رادیکال های آزاد و تابش اشعه ماورا بنفش فعال شود. 138 فسفوریلاسیون IkB باعث آزاد شدن NF-kB می شود و اجازه می دهد تا برای فعال سازی رونویسی ژن وارد هسته شود. 139 تعدادی از کینازها در بقایای سرین به IkB های فسفریلات گزارش شده است. این کینازها اهداف سیگنال های اکسیداتیو برای فعال سازی NF-kB.140 هستند. عوامل کاهنده اتصال DNA NF-kB را افزایش می دهند ، در حالی که عوامل اکسید کننده مانع اتصال DNA NF-kB می شوند. TRX ممکن است 2 عمل مخالف در تنظیم NF-kB اعمال کند: در سیتوپلاسم ، تخریب IkB را مسدود کرده و از فعالیت NF-kB جلوگیری می کند اما اتصال DNA NF-kB در هسته را افزایش می دهد .141 فعال سازی NF-kB از طریق تخریب مربوط به اکسیداسیون نتایج IkB منجر به فعال شدن چندین ژن مرتبط با دفاع آنتی اکسیدانی می شود. NF-kB بیان چندین ژن را که در پاسخ ایمنی شرکت می کنند تنظیم می کند ، مانند IL-1b ، IL-6 ، فاکتور نکروز تومور-a ، IL-8 ، و چندین مولکول چسبندگی. 142,143،XNUMX NF-kB همچنین آنژیوژنز و تکثیر را تنظیم می کند و تمایز سلول ها.
AP-1 نیز توسط دولت ردوکس تنظیم می شود. در حضور H2O2، برخی از یونهای فلزی می توانند از AP-1 فعال شوند. افزایش نسبت GSH / GSSG افزایش اتصال AP-1 در حالی که GSSG مهار اتصال DNA اتصال AP-1.144 DNA از Fos / ژن heterodimer توسط کاهش یک سیتستین محافظت شده تک در دامنه اتصال DNA هر یک از پروتئین های 145 در حالی که اتصال DNA از AP-1 توسط GSSG در بسیاری از انواع سلول ها مهار می شود، نشان می دهد که ایجاد پیوند دیسفالید توسط بقایای cysteine باعث مهار اتصال DNA AP-1 می شود. 146,147 انتقال سیگنال از طریق استرس اکسیداتیو در شکل 2 خلاصه می شود.
نتیجه گیری
استرس اکسیداتیو می تواند در اثر تولید بیش از حد ROS بوسیله واکنش های متابولیکی که از اکسیژن استفاده می کنند و تعادل بین آنها را تغییر دهد اکسیدان /آنتی اکسیدان وضعیت به نفع اکسیدان ها. ROS توسط فعالیت های متابولیک سلولی و عوامل محیطی مانند آلاینده های هوا یا سیگار تولید می شود. ROS مولکول های بسیار واکنشی هستند زیرا الکترون های نامشخص در ساختار آنها هستند و با چندین ماکرومولکول بیولوژیکی در سلول مانند کربوهیدرات ها، اسیدهای نوکلئیک، لیپید ها و پروتئین ها واکنش می دهند و عملکرد آنها را تغییر می دهند. ROS بر بیان ژن های مختلف با تنظیم مقادیر حساس ردکس حساس و ترمیم کروماتین با تغییر در استیلاسیون / دیاستیل کردن هیستون تأثیر می گذارد. مقررات مربوط به حالت رادیواکتیو برای زنده ماندن، فعال شدن، تکثیر و عملکرد اندام حیاتی است.
مراجع
1. Valko M، Rhodes CJ، Moncol J، Izakovic M، Mazur M. رادیکال های آزاد ، فلزات و آنتی اکسیدان ها در سرطان ناشی از استرس اکسیداتیو. Chem Biol Interact. 2006 ؛ 160: 1 40.
2 Halliwell B، Gutteridge JMC. رادیکال های آزاد در زیست شناسی و پزشکی. 3rd ed. نیویورک: انتشارات دانشگاه آکسفورد، 1999.
3. مارنت LJ. پراکسیداسیون لیپید آسیب dDNA توسط مالون دی آلدئید. Mutat Res. 1999 ؛ 424: 83 95.
4. تشکیل Siems WG ، Grune T ، Esterbauer H. 4-Hydroxynonenal در طی ایسکمی و خونرسانی مجدد روده کوچک موش. ifeعلم زندگی. 1995 ؛ 57: 785 789.
5. Stadtman ER. نقش گونه های اکسیدان در پیری. Curr Med شیمی. 2004 ؛ 11: 1105 1112.
6. Wang MY ، Dhingra K ، Hittelman WN ، Liehr JG ، deAndrade M ، Li DH. ترکیبات افزایشی مالون دی آلدئید ativeDNA ناشی از پراکسیداسیون لیپیدها در بافتهای پستان انسان نشانگرهای زیستی اپیدمیول سرطان 1996 ؛ 5: 705 710.
7. Jenner P. استرس اکسیداتیو در بیماری پارکینسون. آن نورول 2003 ؛ 53: S26 S36.
8. Lyras L، Cairns NJ، Jenner A، Jenner P، Halliwell B. ارزیابی آسیب اکسیداتیو به پروتئین ها ، لیپیدها و DNA در مغز بیماران مبتلا به بیماری آلزایمر. جی نوروشیم. 1997 ؛ 68: 2061 2069.
9. Sayre LM ، Smith MA ، Perry G. شیمی و بیوشیمیایی استرس اکسیداتیو در بیماری تخریب عصبی. Curr Med شیمی. 2001 ؛ 8: 721 738.
10. Toshniwal PK ، Zarling EJ. شواهدی در مورد افزایش پراکسیداسیون لیپید در بیماری ام اس. Neurochem Res. 1992 ؛ 17: 205 207.
11. Dhalla NS، Temsah RM، Netticadan T. نقش استرس اکسیداتیو در بیماری های قلبی عروقی. هایپرتنس 2000 ؛ 18: 655-673.
12. Kasparova S ، Brezova V ، Valko M ، Horecky J ، Mlynarik V ، و دیگران. مطالعه استرس اکسیداتیو در مدل موش صحرایی پرفیوژن مزمن. Neurochem Int. 2005 ؛ 46: 601 611.
13. Kerr S، Brosnan MJ، McIntyre M، Reid JL، Dominiczak AF، Hamilton CA. تولید آنیون سوپراکسید در یک مدل فشار خون ژنتیکی افزایش می یابد: نقش اندوتلیوم. فشار خون. 1999 ؛ 33: 1353 1358.
14. Kukreja RC ، Hess ML. سیستم رادیکال های آزاد اکسیژن: از معادلات از طریق فعل و انفعالات غشا پروتئین گرفته تا آسیب و محافظت در قلب و عروق. Cardiovasc Res. 1992 ؛ 26: 641 655.
15. Asami S، Manabe H، Miyake J، Tsurudome Y، Hirano T، et al. سیگار کشیدن باعث افزایش آسیب اکسیداتیو DNA ، 8-هیدروکسی دئوکسی گوانوزین ، در یک محل مرکزی ریه انسان می شود. سرطان زایی 1997 ؛ 18: 1763 1766.
16. Andreadis AA ، Hazen SL ، Comhair SA ، ارزروم SC. رویدادهای اکسیداتیو و نیتروزاتیو در آسم. رایگان Radic Biol Med. 2003 ؛ 35: 213 225.
17. Comhair SA ، Ricci KS ، Arroliga M ، Lara AR ، Dweik RA ، و دیگران. همبستگی کمبود سوپراکسید دیسموتاز سیستمیک با انسداد جریان هوا در آسم. Am J Respir Crit Care Med. 2005 ؛ 172: 306 313.
18. Comhair SA ، Xu W ، Ghosh S ، Thunnissen FB ، Almasan A ، و دیگران. غیرفعال سازی سوپراکسید دیسموتاز در پاتوفیزیولوژی بازسازی راه هوایی آسم و واکنش آن Am J Pathol. 2005 ؛ 166: 663-674.
19. Dut R ، Dizdar EA ، Birben E ، Sackesen C ، Soyer OU ، Besler T ، Kalayci O. استرس اکسیداتیو و عوامل تعیین کننده آن در مجاری تنفسی کودکان مبتلا به آسم. آلرژی 2008 ؛ 63: 1605 1609.
20. Ercan H ، Birben E ، Dizdar EA ، Keskin O ، Karaaslan C ، و دیگران. استرس اکسیداتیو و عوامل تعیین کننده ژنتیکی و اپیدمیولوژیک آسیب اکسیدان در آسم در کودکان J آلرژی کلینیک ایمونول. 2006 ؛ 118: 1097 1104.
21. Fitzpatrick AM، Teague WG، Holguin F، Yeh M، Brown LA. برنامه تحقیقاتی آسم شدید. هموستاز گلوتاتیون راه هوایی در کودکان مبتلا به آسم شدید تغییر می کند: شواهدی برای استرس اکسیدان. J آلرژی کلینیک ایمونول. 2009 ؛ 123: 146 152.
22. Miller DM، Buettner GR، Aust SD. فلزات انتقالی به عنوان کاتالیزور واکنش های "اکسیداسیون". رایگان Radic Biol Med. 1990 ؛ 8: 95 108.
23. Dupuy C ، Virion A ، Ohayon R ، Kaniewski J ، D me D ، Pommier J. مکانیسم تشکیل پراکسید هیدروژن توسط NADPH اکسیداز در غشای پلاسمای تیروئید کاتالیز شده است. J Biol شیمی. 1991 ؛ 266: 3739 3743.
24. گرنجر DN. نقش گزانتین اکسیداز و گرانولوسیت در آسیب ایسکمی پرفیوژن Am J Physiol. 1988 ؛ 255: H1269 H1275.
25. Fenton HJH. اکسیداسیون اسید تارتاریک در حضور آهن. J Chem Soc. 1984 ؛ 65: 899 910.
26. Haber F ، Weiss JJ. تجزیه کاتالیستی پراکسید هیدروژن توسط نمک های آهن. Proc R Soc Lond Ser A. 1934 ؛ 147: 332 351.
27. Liochev SI ، فریدویچ I. Haber Weiss 70 سال بعد دوچرخه سواری کرد: یک دیدگاه جایگزین. Redox Rep. 2002 ؛ 7: 55 57.
28. Klebanoff SJ. میلوپراکسیداز: دوست و دشمن. J Leukoc Biol. 2005 ؛ 77: 598 625.
29. Whiteman M ، Jenner A ، Halliwell B. تغییرات پایه ناشی از اسید هیپوکلروس در DNA تیموس گوساله جدا شده. شیمی Res Toxicol. 1997 ؛ 10: 1240 1246.
30. Kulcharyk PA، Heinecke JW. اسید هیپوکلروس تولید شده توسط سیستم میلوپراکسیداز فاگوسیت های انسانی باعث ایجاد پیوندهای متقابل کووالانسی بین DNA و پروتئین می شود. بیوشیمی. 2001 ؛ 40: 3648-3656.
31. Brennan ML، Wu W، Fu X، Shen Z، Song W، et al. داستان دو بحث: تعریف هر دو نقش پراكسیدازها در ایجاد نیتروتیروزین در داخل بدن با استفاده از ائوزینوفیل پراكسیداز و موشهای دارای مایلوپراكسیداز و همچنین ماهیت گونههای نیتروژن واكنش دار تولید پراكسیداز J Biol شیمی. 2002 ؛ 277: 17415 17427.
32. Denzler KL، Borchers MT، Crosby JR، Cieslewicz G، Hines EM، et al. دگرانولاسیون گسترده ائوزینوفیل و اکسیداسیون واسطه پروکسیداز پروتئین های مجاری هوایی در یک مدل التهاب ریوی با چالش اووالبومین موش رخ نمی دهد. ج ایمونول. 2001 ؛ 167: 1672 1682.
33. van Dalen CJ، Winterbourn CC، Senthilmohan R، Kettle AJ. نیتریت به عنوان یک بستر و مهار کننده میلوپراکسیداز. پیامدهای تولید نیترات و اسید هیپوکلروس در مکان های التهاب J Biol شیمی. 2000 ؛ 275: 11638 11644.
34. Wood LG، Fitzgerald DA، Gibson PG، Cooper DM، Garg ML. پراکسیداسیون لیپیدها توسط ایزوپروستان های پلاسما تعیین می شود به شدت بیماری در آسم خفیف مربوط است. چربی ها 2000 ؛ 35: 967 974.
35. Montuschi P ، Corradi M ، Ciabattoni G ، Nightingale J ، Kharitonov SA ، Barnes PJ. افزایش 8 ایزوپروستان ، نشانگر استرس اکسیداتیو ، در میعانات بازدم بیماران آسم. Am J Respir Crit Care Med. 1999 ؛ 160: 216 220.
36. Church DF، Pryor WA. شیمی رادیکال های آزاد دود سیگار و پیامدهای سم شناسی آن. چشم انداز بهداشت محیط. 1985 ؛ 64: 111 126.
37. Hiltermann JT، Lapperre TS، van Bree L، Steerenberg PA، Brahim JJ، et al. التهاب ناشی از ازن در خلط و مایع شستشوی برونش از افراد مبتلا به آسم ارزیابی شده است: یک ابزار غیرتهاجمی جدید در مطالعات اپیدمیولوژیک در مورد آلودگی هوا و آسم رایگان Radic Biol Med. 1999 ؛ 27: 1448-1454.
38. Nightingale JA ، Rogers DF ، بارنز PJ. تأثیر ازن استنشاق شده بر روی اکسید نیتریک بازدم ، عملکرد ریوی و خلط ناشی از آن در افراد طبیعی و آسم. قفسه سینه 1999 ؛ 54: 1061 1069.
39. Cho AK، Sioutas C، Miguel AH، Kumagai Y، Schmitz DA، et al. فعالیت Redox ذرات معلق در هوا در سایت های مختلف حوزه لس آنجلس. محیط زیست 2005 ؛ 99: 40 47.
40. Comhair SA ، Thomassen MJ ، ارزروم SC. القای افتراقی گلوتاتیون پراکسیداز خارج سلولی و نیتریک اکسید سنتاز 2 در مجاری هوایی افراد سالم که در معرض 100٪ O (2) یا دود سیگار هستند. Am J Respir Cell Mol Biol. 2000 ؛ 23: 350 354.
41. Matthay MA، Geiser T، Matalon S، Ischiropoulos H. آسیب ریه با واسطه اکسیدان در سندرم دیسترس حاد تنفسی. Crit Care Med. 1999 ؛ 27: 2028 2030.
42. Biaglow JE ، Mitchell JB ، Held K. اهمیت پراکسید و سوپراکسید در پاسخ اشعه X. Int J Radiat Oncol Biol Phys. 1992 ؛ 22: 665 669.
43. Chiu SM ، Xue LY ، Friedman LR ، Oleinick NL. حساسیت یونی مس به سایت های اتصال ماتریس هسته ای به اشعه یونیزان. بیوشیمی. 1993 ؛ 32: 6214 6219.
44. Narayanan PK ، Goodwin EH ، Lehnert BE. ذرات آلفا تولید بیولوژیکی آنیونهای سوپراکسید و پراکسید هیدروژن در سلولهای انسانی را آغاز می کنند. سرطان رز 1997 ؛ 57: 3963 3971.
45. Tuttle SW، Varnes ME، Mitchell JB، Biaglow JE. حساسیت به اکسیدان های شیمیایی و تابش در رده های سلولی CHO با کمبود فعالیت چرخه پنتوز اکسیداتیو Int J Radiat Oncol Biol Phys. 1992 ؛ 22: 671 675.
46 Guo G، Yan-Sanders Y، Lyn-Cook BD، Wang T، Tamae D، et al. منگنز
بیان ژن به وسیله سوپراکسید دیسموتاز در تشعشع اشعه
پاسخ های سازگار مول سلول بیول. 2003 ؛ 23: 2362 2378.
47 آزام EI، د تولدو SM، Spitz DR، Little JB. متابولیسم اکسیداتیو
مدولاسیون انتقال سیگنال و تشکیل میکرونی در سمت راست است
سلول های فیبروبلاست های طبیعی بدن انسان را تابش می کند. سرطان Res.
2002 ؛ 62: 5436 5442.
48 لیچ جی. ک.، ون توایل گ، لینس PS، اشمیت الیچر، ربک میککلزن.
مولکول واکنش پذیر وابسته به میتوکندری ناشی از تابش یونیزه شده است
اکسیژن / نیتروژن. سرطان رز 2001 ؛ 61: 3894-3901.
49 Dent P، Yacoub A، Fisher PB، MP Hagan، Grant S. مسیر MAPK در
پاسخ های تابش انکوژن 2003 ؛ 22: 5885 5896.
50 Wei SJ، Botero A، Hirota K، Bradbury CM، Markovina S، و همکاران. تیرودوکسین
جابجایی هسته ای و تعامل با فاکتور ردوکس -1 عامل رونویسی AP-1 را در پاسخ به تابش یونیزان فعال می کند. سرطان رز 2000 ؛ 60: 6688 6695.
51. Cadet J ، Douki T ، Gasparutto D ، Ravanat JL. آسیب اکسیداتیو به DNA: شکل گیری ، اندازه گیری و ویژگی های بیوشیمیایی. Mutat Res. 2003 ؛ 531: 5 23.
52. Yokoya A، Cunniffe SM، O Neill P. اثر هیدراتاسیون در القای شکستگی رشته ها و ضایعات پایه در فیلم های DNA پلاسمید توسط gammaradiation. J Am Chem Soc. 2002 ؛ 124: 8859 8866.
53. Janssen YM، Van Houten B، Borm PJ، Mossman BT. پاسخ سلول و بافت به آسیب اکسیداتیو. آزمایشگاه سرمایه گذاری 1993 ؛ 69: 261 274.
54. Iwanaga M ، Mori K ، Iida T ، Urata Y ، Matsuo T ، و دیگران. فاکتور هسته ای القا ka وابسته به گاما گلوتامیل سیستئین سنتتاز با اشعه یونیزه در سلول های گلیوبلاستومای انسانی T98G رایگان Radic Biol Med. 1998 ؛ 24: 1256 1268.
55. Stohs SJ، Bagchi D. مکانیسم های اکسیداتیو در سمیت یون های فلزی. رایگان Radic Biol Med. 1995 ؛ 18: 321 336.
56. Leonard SS، Harris GK، Shi X. استرس اکسیداتیو ناشی از فلز و انتقال سیگنال. رایگان Radic Biol Med. 2004 ؛ 37: 1921 1942.
57. Shi H ، Shi X ، Liu KJ. مکانیسم اکسیداتیو سمیت آرسنیک و سرطان زایی Mol Cell Biochem. 2004 ؛ 255: 67 78.
58. Pi J، Horiguchi S، Sun Y، Nikaido M، Shimojo N، Hayashi T. مکانیسم بالقوه ای برای اختلال در تشکیل اکسید نیتریک ناشی از قرار گرفتن طولانی مدت دهان به آرسنات در خرگوش ها. Free Radic Biol Med. 2003 ؛ 35: 102 113.
59. Rin K ، Kawaguchi K ، Yamanaka K ، Tezuka M ، Oku N ، Okada S. استراحت DNA استرند ناشی از اسید دی متیلارزینیک ، متابولیتی از مواد زائد غیر آلی ، توسط رادیکال های آنیون سوپراکسید به شدت تقویت می شود. Biol Pharm Bull. 1995 ؛ 18: 45 58.
60. Waalkes MP، Liu J، Ward JM، Diwan LA. مکانیسم های زمینه ساز سرطان آرسنیک: حساسیت بیش از حد موش های در معرض آرسنیک غیر آلی در دوران بارداری سم شناسی. 2004 ؛ 198: 31 38.
61. Schiller CM، Fowler BA، Woods JS. اثرات آرسنیک بر فعال سازی پیروات دهیدروژناز چشم انداز بهداشت محیط. 1977 ؛ 19: 205 207.
62. Monterio HP ، Bechara EJH ، Abdalla DSP. درگیری رادیکال های آزاد در پورفیریای عصبی و مسمومیت با سرب. Mol Cell Biochem. 1991 ؛ 103: 73 83.
63. Tripathi RM ، Raghunath R ، Mahapatra S. سرب خون و تأثیر آن بر میزان Cd ، Cu ، Zn ، Fe و هموگلوبین کودکان. Sci Total Environ. 2001 ؛ 277: 161 168.
64. Nehru B، Dua R. اثر سلنیوم در رژیم غذایی بر مسمومیت عصبی سرب. J Environ Pathol Toxicol Oncol. 1997 ؛ 16: 47 50.
65. Reid TM، Feig DI، Loeb LA. جهش زایی توسط رادیکال های اکسیژن ناشی از فلز. چشم انداز بهداشت محیط. 1994 ؛ 102 (مکمل 3): 57 61.
66. Kinnula VL، Crapo JD. سوپراکسید دیسموتازها در بیماری های ریه و انسان. Am J Respir Crit Care Med. 2003 ؛ 167: 1600 1619.
67. Kinnula VL. تولید و تخریب متابولیت های اکسیژن در طی شرایط التهابی در ریه انسان. داروی Curr آلرژی التهاب را هدف قرار می دهد. 2005 ؛ 4: 465 470.
68. Zelko IN، Mariani TJ، Folz RJ. خانواده چند ژنی سوپراکسید دیسموتاز: مقایسه ساختارهای ژن ، تکامل و بیان ژن CuZn-SOD (SOD1) ، Mn-SOD (SOD2) و EC-SOD (SOD3). رایگان Radic Biol Med. 2002 ؛ 33: 337 349.
69. Kirkman HN ، Rolfo M ، Ferraris AM ، Gaetani GF. مکانیسم های محافظت از کاتالاز توسط NADPH. سینتیک و استوکیومتری. J Biol شیمی. 1999 ؛ 274: 13908 13914.
70. Floh L. گلوتاتیون پراکسیداز. علوم پایه زندگی. 1988 ؛ 49: 663 668.
71. آرتور جونیور گلوتاتیون پراکسیدازها. Cell Mol Life Sci. 2000 ؛ 57: 1825 1835.
72. Chu FF ، Doroshow JH ، Esworthy RS. بیان ، خصوصیات و توزیع بافتی یک گلوتاتیون پراکسیداز وابسته به سلنیوم سلولی ، GSHPx-GI. J Biol شیمی. 1993 ؛ 268: 2571 2576.
73. Comhair SA ، Bhathena PR ، Farver C ، Thunnissen FB ، Erzurum SC. القای گلوتاتیون پراکسیداز خارج سلولی در ریه های آسم: شواهدی برای تنظیم اکسایش اکسیداسیون در سلول های اپیتلیال مجاری تنفسی انسان FASEB J. 2001 ؛ 15: 70-78.
74. Gromer S، Urig S، Becker K. سیستم thioredoxin از علم به کلینیک. Med Res Rev. 2004 ؛ 24: 40-89.
75. Kinnula VL ، Lehtonen S ، Kaarteenaho-Wiik R ، Lakari E ، P kk P ، و دیگران. بیان اختصاصی سلول پراکسیروکسین در ریه انسان و سارکوئیدوز ریوی قفسه سینه 2002 ؛ 57: 157 164.
76. Dubuisson M ، Vander Stricht D ، Clippe A ، Etienne F ، Nauser T ، و دیگران. پراکسیریدوکسین 5 انسان یک پروکسینیتریت ردوکتاز است. FEBS Lett. 2004 ؛ 571: 161 165.
77. Holmgren A. عملکرد آنتی اکسیدانی سیستم های تیوردوکسین و گلوتاردوکسین. سیگنال آنتی اکسید ردوکس. 2000 ؛ 2: 811 820.
78. دیکینسون DA ، Forman HJ. گلوتاتیون در دفاع و سیگنالینگ: درسهایی از یک تیول کوچک. Ann NY Acad Sci. 2002 ؛ 973: 488-504.
79. Sies H. گلوتاتیون و نقش آن در عملکردهای سلولی. رایگان Radic Biol Med. 1999 ؛ 27: 916 921.
80. Ladner JE ، Parsons JF ، Rife CL ، Gilliland GL ، Armstrong RN. مسیرهای تکاملی موازی برای ترانسفرازهای گلوتاتیون: ساختار و مکانیسم آنزیم کاپا rGSTK1-1 کلاس میتوکندری بیوشیمی. 2004 ؛ 43: 52 61.
81. Robinson A، Huttley GA، Booth HS، Board PG. مطالعات مدلسازی و بیوانفورماتیک از کلاس گلوتاتیون ترانسفراز کلاس کاپا ، یک خانواده سوم ترانسفراز جدید را با همسانی به ایزومرازهای 2-هیدروکسی کرومن-2-کربوکسیلات پروکاریوتی پیش بینی می کند. Biochem J. 2004 ؛ 379: 541-552.
82. Jakobsson PJ، Morgenstern R، Mancini J، Ford-Hutchinson A، Persson B. ویژگیهای ساختاری متداول MAPEGda ابرخانواده گسترده پروتئینهای مرتبط با غشا با عملکردهای بسیار واگرا در متابولیسم eicosanoid و گلوتاتیون. پروتئین علمی 1999 ؛ 8: 689-692.
83. Hayes JD ، Pulford DJ. خانواده سوپرگن گلوتاتیون S-ترانسفراز: تنظیم GST و سهم ایزوآنزیم ها در محافظت شیمیایی و مقاومت دارویی سرطان Crit Rev Biochem Mol Biol. 1995 ؛ 30: 445 600.
84. آرمسترانگ RN. ساختار ، مکانیسم کاتالیزوری و تکامل ترانسفرازهای گلوتاتیون. شیمی Res Toxicol. 1997 ؛ 10: 2 18.
85. Hayes JD، McLellan LI. آنزیم های گلوتاتیون و گلوتاتیون وابسته نشان دهنده یک دفاع هماهنگ تنظیم شده در برابر استرس اکسیداتیو هستند. رایگان Radic Res. 1999 ؛ 31: 273 300.
86. Sheehan D ، Meade G ، Foley VM ، Dowd CA. ساختار ، عملکرد و تکامل ترانسفرازهای گلوتاتیون: پیامدهای طبقه بندی اعضای غیر پستانداران یک خانواده آنزیم باستانی Biochem J. 2001 ؛ 360: 1 16.
87. Cho SG، Lee YH، Park HS، Ryoo K، Kang KW، et al. گلوتاتیون S-ترانسفراز Mu با سرکوب کیناز تنظیم کننده سیگنال آپوپتوز ، سیگنالهای فعال شده در استرس را تعدیل می کند. J Biol Chem. 1 ؛ 2001: 276 12749.
88. Dorion S، Lambert H، Landry J. فعال سازی مسیر سیگنالینگ p38 توسط شوک حرارتی شامل تفکیک گلوتاتیون S- ترانسفراز Mu از Ask1 است. J Biol شیمی. 2002 ؛ 277: 30792 30797.
89. Adler V، Yin Z، Fuchs SY، Benezra M، Rosario L، et al. تنظیم سیگنالینگ JNK توسط GSTp. EMBO J. 1999 ؛ 18: 1321 1334.
90. Manevich Y ، Feinstein SI ، Fisher AB. فعال سازی آنزیم آنتی اکسیدان 1-CYS پراکسیریدوکسین نیاز به گلوتاتیونیلاسیون با واسطه هترو دیمریزاسیون با pGST دارد. Proc Natl Acad Sci US A. 2004 ؛ 101: 3780 3785.
91. بنکر VW. رادیکال های آزاد ، آنتی اکسیدان ها و پیری. علوم آزمایشگاهی Med. 1992 ؛ 49: 299-312.
92. Mezzetti A ، Lapenna D ، Romano F ، Costantini F ، Pierdomenico SD ، و دیگران. استرس اکسیداتیو سیستمیک و ارتباط آن با سن و بیماری. J Am Geriatr Soc. 1996 ؛ 44: 823 827.
93 سفید E، شانون JS، Patterson RE. رابطه بین ویتامین و
استفاده از مکمل کلسیم و سرطان روده بزرگ. نشانگرهای زیستی اپیدمیول سرطان 1997 ؛ 6: 769 774.
94. Masella R، Di Benedetto R، Vari R، Filesi C، Giovannini C. مکانیسم های جدید ترکیبات آنتی اکسیدانی طبیعی در سیستم های بیولوژیکی: درگیری آنزیم های مربوط به گلوتاتیون و گلوتاتیون. J Nutr Biochem. 2005 ؛ 16: 577 586.
95. Curello S ، Ceconi C ، Bigoli C ، Ferrari R ، Albertini A ، Guarnieri C. تغییرات در وضعیت گلوتاتیون قلب پس از ایسکمی و خونرسانی مجدد. تجربه 1985 ؛ 41: 42 43.
96. El-Agamey A ، Lowe GM ، McGarvey DJ ، Mortensen A ، Phillip DM ، Truscott TG. شیمی رادیکال کاروتنوئید و خواص آنتی اکسیدانی / پرو اکسیدانی. Arch Biochem Biophys. 2004 ؛ 430: 37 48.
97. Rice-Evans CA، Sampson J، Bramley PM، Holloway DE. چرا انتظار داریم کاروتنوئیدها در داخل بدن آنتی اکسیدان باشند؟ رایگان Radic Res. 1997 ؛ 26: 381 398.
98. Niles RM. مسیرهای سیگنالینگ در پیشگیری شیمیایی رتینوئید و درمان سرطان Mutat Res. 2004 ؛ 555: 81 96.
99. Donato LJ، Noy N. سرکوب رشد کارسینومای پستانی توسط اسید رتینوئیک: ژن های پرواپوپتوتیک اهدافی برای گیرنده های اسید رتینوئیک و سیگنالینگ پروتئین II متصل به اسید رتینوئیک سلول هستند. سرطان رز 2005 ؛ 65: 8193 8199.
100. Niizuma H ، Nakamura Y ، Ozaki T ، Nakanishi H ، Ohira M ، و دیگران. Bcl-2 یک تنظیم کننده اصلی برای مرگ سلولهای آپوپتوز ناشی از اسید رتینوئیک در نوروبلاستوما است. انکوژن 2006 ؛ 25: 5046 5055.
101. Dalton TP، Shertzer HG، Puga A. تنظیم بیان ژن توسط اکسیژن واکنش پذیر. Ann Rev Pharmacol Toxicol. 1999 ؛ 39: 67 101.
102. Scandalios JG. پاسخ های ژنومی به استرس اکسیداتیو. در: Meyers RA ، ed. دانشنامه زیست شناسی سلول های مولکولی و پزشکی مولکولی. جلد 5. ویرایش دوم. وینهایم ، آلمان: Wiley-VCH؛ 2: 2004 489.
103. Ghosh R، Mitchell DL. تأثیر آسیب اکسیداتیو DNA در عناصر پروموتر بر اتصال فاکتور رونویسی Nucleic Acids Res. 1999 ؛ 27: 3213 3218.
104. Marietta C، Gulam H، Brooks PJ. یک ضایعه منفرد 8 ، 50 سیکلو -20-دئوکسیادنوزین در یک جعبه TATA از اتصال پروتئین متصل به TATA جلوگیری می کند و رونویسی را در داخل بدن به شدت کاهش می دهد. ترمیم DNA (Amst). 2002 ؛ 1: 967 975.
105 جکسون AL، چن R، Loeb LA. القای بی ثباتی ریزماهواره
توسط آسیب اکسیداتیو DNA. Proc Natl Acad Sci US A. 1998 ؛ 95: 12468 12473.
106. Caldecott KW. فعل و انفعالات پروتئین و پروتئین در طول ترمیم شکستگی تک رشته DNA پستانداران. Biochem Soc Trans. 2003 ؛ 31: 247 251.
107. Cooke MS، Evans MD، Dizdaroglu M، Lunec J. آسیب DNA اکسیداتیو: مکانیسم ها ، جهش و بیماری. FASEB J. 2003 ؛ 17: 1195 1214.
108. Jones PL، Wolffe AP. روابط بین سازمان کروماتین و متیلاسیون DNA در تعیین بیان ژن. Semin Cancer Biol. 1999 ؛ 9: 339 347.
109. Girotti AW. مکانیسم پراکسیداسیون لیپیدها. J Free Radic Biol Med. 1985 ؛ 1: 87 95.
110. Siu GM ، Draper HH. متابولیسم مالون آلدئید در داخل بدن و in vitro. لیپیدها 1982 ؛ 17: 349 355.
111. Esterbauer H، Koller E، Slee RG، Koster JF. درگیری احتمالی محصول پراکسیداسیون لیپید 4-هیدروکسی زنا در تشکیل کرومولیپیدهای فلورسنت. Biochem J. 1986 ؛ 239: 405 409.
112. Hagihara M ، Nishigaki I ، Maseki M ، Yagi K. تغییرات وابسته به سن در سطح پراکسید لیپید در بخشهای لیپوپروتئین سرم انسان. ج Gerontol. 1984 ؛ 39: 269 272.
113. Keller JN، Mark RJ، Bruce AJ، Blanc E، Rothstein JD، et al. 4- Hydroxynonenal ، یک محصول آلدئیدی پراکسیداسیون لیپیدهای غشایی ، حمل و نقل گلوتامات و عملکرد میتوکندری را در سیناپتوزوم ها مختل می کند. علوم اعصاب 1997 ؛ 806: 85 96.
114. Uchida K، Shiraishi M، Naito Y، Torii Y، Nakamura Y، Osawa T. فعال سازی مسیرهای سیگنالینگ استرس توسط محصول نهایی پراکسیداسیون لیپید. 4-هیدروکسی-2-نونال یک القا potential کننده بالقوه تولید پراکسید داخل سلول است. J Biol شیمی. 1999 ؛ 274: 2234 2242.
115. Suc I ، Meilhac O ، Lajoie-Mazenc I ، Vandaele J ، Jurgens G ، Salvayre R ، Negre-Salvayre A. فعال سازی گیرنده EGF توسط LDL اکسید شده. FASEB J. 1998 ؛ 12: 665 671.
116. Tsukagoshi H ، Kawata T ، Shimizu Y ، Ishizuka T ، Dobashi K ، Mori M. 4-Hydroxy-2-nonenal تولید فیبرونکتین توسط فیبروبلاست های ریه انسان IMR-90 را تا حدی از طریق فعال سازی سیگنال خارج سلولی متصل به گیرنده فاکتور رشد اپیدرم افزایش می دهد- مسیر کیناز تنظیم شده p44 / 42. Toxicol Appl Pharmacol. 2002 ؛ 184: 127 135.
117. Montuschi P ، Collins JV ، Ciabattoni G ، Lazzeri N ، Corradi M ، Kharitonov SA ، Barnes PJ. 8-ایزوپروستان را به عنوان بیومارکر استرس اکسیداتیو ریه در بیماران داخل ریه و سیگاری های سالم بازدم می کنید. Am J Respir Crit Care Med. 2000 ؛ 162: 1175 1177.
118. موریسون D ، رحمان اول ، Lannan S ، MacNee W. نفوذپذیری اپیتلیال ، التهاب و استرس اکسیدان در فضای هوای افراد سیگاری. Am J Respir Crit Care Med. 1999 ؛ 159: 473 479.
119. Nowak D ، Kasielski M ، Antczak A ، Pietras T ، Bialasiewicz P. افزایش محتوای مواد واکنش دهنده اسید تیوباربیتوریک و پراکسید هیدروژن در میعانات تنفسی منقضی شده بیماران با بیماری انسداد ریوی مزمن پایدار: اثر قابل توجهی در استعمال سیگار نیست. رسپیر مد 1999 ؛ 93: 389 396.
120. کلی FJ ، Mudway IS. اکسیداسیون پروتئین در رابط هوا و ریه. آمینو اسید. 2003 ؛ 25: 375 396.
121. Dean RT ، Roberts CR ، Jessup W. تقسیم پلی پپتیدهای خارج سلولی و داخل سلولی توسط رادیکال های آزاد. Prog Clin Biol Res. 1985 ؛ 180: 341 350.
122. کک RG. استفاده از هیدروپراکسید t-butyl به عنوان پروب اکسیداسیون متیونین در پروتئین ها. بیوشیم مقعدی 1996 ؛ 236: 56 62.
123. دیویس کی جی تخریب و تخریب پروتئین توسط رادیکال های اکسیژن. I. جنبه های کلی. J Biol شیمی. 1987 ؛ 262: 9895 9901.
124 Stadtman ER اکسیداسیون یونهای کاتالیزوری پروتئین: مکانیزم بیوشیمیایی و پیامدهای بیولوژیکی. رایگان Radic Biol Med.
1990 ؛ 9: 315 325.
125. Fucci L ، Oliver CN ، Coon MJ ، Stadtman ER. غیرفعال سازی آنزیم های مهم متابولیکی توسط واکنش های اکسیداسیون با عملکرد مخلوط: پیامدهای احتمالی در گردش پروتئین و پیری Proc Natl Acad Sci US A. 1983 ؛ 80: 1521 1525.
126. Stadtman ER ، Moskovitz J ، Levine RL. اکسیداسیون بقایای متیونین پروتئین ها: عواقب بیولوژیکی سیگنال آنتی اکسید ردوکس. 2003 ؛ 5: 577 582.
127. Stadtman ER ، Levine RL. اکسیداسیون اسیدهای آمینه آزاد و باقیمانده اسیدهای آمینه با واسطه رادیکال های آزاد در پروتئین ها. آمینو اسید. 2003 ؛ 25: 207 218.
128. Stadtman ER. اکسیداسیون پروتئین در پیری و بیماری های وابسته به سن. Ann NY Acad Sci. 2001 ؛ 928: 22 38.
129. Shacter E. کمیت و اهمیت اکسیداسیون پروتئین در نمونه های بیولوژیکی. Drug Metab Rev. 2000 ؛ 32: 307 326.
130. Poli G ، Leonarduzzi G ، Biasi F ، Chiarpotto E. استرس اکسیداتیو و سیگنالینگ سلولی. Curr Med شیمی. 2004 ؛ 11: 1163 1182.
131. Neufeld G، Cohen T، Gengrinovitch S، Poltorak Z. فاکتور رشد اندوتلیال عروقی (VEGF) و گیرنده های آن. FASEB J. 1999 ؛ 13: 9 22.
132. Sundaresan M ، Yu ZX ، Ferrans VJ ، Sulciner DJ ، Gutkind JS ، و دیگران. تنظیم تولید گونه های اکسیژن واکنش پذیر در فیبروبلاست ها توسط Rac1. Biochem J. 1996 ؛ 318: 379-382.
133. Sun T ، Oberley LW. تنظیم Redox فعال کننده های رونویسی. رایگان Radic Biol Med. 1996 ؛ 21: 335 348.
134. Klatt P ، Molina EP ، De Lacoba MG ، Padilla CA ، Martinez-Galesteo E ، Barcena JA ، Lamas S. Redox تنظیم اتصال DNA c-Jun توسط S-glutathiolation برگشت پذیر. FASEB J. 1999 ؛ 13: 1481 1490.
135 Reynaert NL، Ckless K، Guala AS، Wouters EF، van der Vliet A، Janssen Heininger
YM در محل تشخیص پروتئین های S-glutathionylated زیر glutaredoxin-1 کاتالیز مشتق سیستئین. Biochim Biophys Acta. 2006 ؛ 1760: 380 387.
136 Reynaert NL، Wouters EF، Janssen-Heininger YM. مدولاسیون glutaredoxin-1
بیان در مدل موش از بیماری راه هوایی آلرژیک - سایپرز ، باشگاه دانش Am J Respir Cell Mol Biol. 2007 ؛ 36: 147 151.
137. Filomeni G ، Rotilio G ، Ciriolo MR. سیگنالینگ سلول و سیستم ردوکس گلوتاتیون. Biochem Pharmacol. 2002 ؛ 64: 1057 1064.
138. Pande V، Ramos MJ. شناخت مولکولی 15-deoxydelta (12,14،2) prostaglandin J (2005) توسط فاکتور هسته ای-کاپا B و سایر پروتئین های سلولی. Bioorg Med Chem Lett. 15 ؛ 4057: 4063 XNUMX.
139. پرکینز ND. ادغام مسیرهای سیگنالینگ سلول با عملکرد NF-kappaB و IKK. Nat Rev Mol Cell Biol. 2007 ؛ 8: 49 62.
140. گیلمور TD. مقدمه ای بر NF-kappaB: بازیکنان ، مسیرها ، دیدگاه ها. انکوژن 2006 ؛ 25: 6680 6684.
141. Hirota K ، Murata M ، Sachi Y ، Nakamura H ، Takeuchi J ، Mori K ، Yodoi J. نقش های متمایز تیوردوکسین در سیتوپلاسم و هسته. یک مکانیسم دو مرحله ای تنظیم ردوکس فاکتور رونویسی NF-kappaB. J Biol شیمی. 1999 ؛ 274: 27891 27897.
142. بخش PA. نقش مکمل ، کموکین ها و سیتوکین های نظارتی در آسیب حاد ریه Ann NY Acad Sci. 1996 ؛ 796: 104 112.
143. Akira S ، Kishimoto A. NF-IL6 و NF-kB در تنظیم ژن سیتوکین. Adv Immunol. 1997 ؛ 65: 1 46.
144. Meyer M، Schreck R، Baeuerle PA. H2O2 و آنتی اکسیدان ها اثرات متضادی در فعال سازی NF-kappa B و AP-1 در سلول های دست نخورده دارند: AP-1 به عنوان یک عامل پاسخ دهنده آنتی اکسیدان ثانویه. EMBO J. 1993 ؛ 12: 2005 2015.
145. Abate C ، Patel L ، Rausher FJ ، Curran T. Redox تنظیم fos و فعالیت اتصال DNA به ژن در شرایط in vitro. علوم پایه. 1990 ؛ 249: 1157 1161.
146. Galter D، Mihm S، Droge W. اثرات متمایز گلوتاتیون دی سولفید در فاکتورهای رونویسی هسته kB و پروتئین فعال کننده 1. Eur J Biochem. 1994 ؛ 221: 639 648.
147. فعالیت های رونویسی Hirota K ، Matsui M ، Iwata S ، Nishiyama A ، Mori K ، Yodoi J. AP-1 توسط ارتباط مستقیم بین تیوردوکسین و Ref-1 تنظیم می شود. Proc Natl Acad Sci US A. 1997 ؛ 94: 3633 3638.
IFM's Find A Practitioner بزرگترین شبکه ارجاع در پزشکی کاربردی است که برای کمک به بیماران در یافتن پزشکان طب عملکردی در هر نقطه از جهان ایجاد شده است. با توجه به تحصیلات گسترده در پزشکی کاربردی ، پزشکان مجاز IFM اولین بار در نتایج جستجو ذکر شده اند