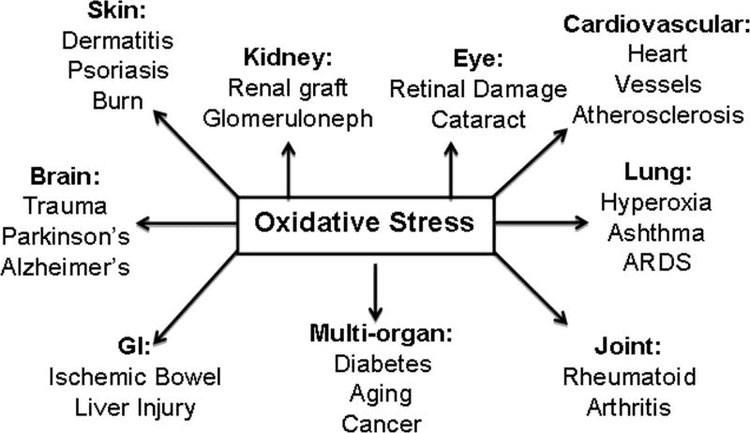
تیم پزشکی عملکردی و کایروپراکتیک استرس اکسیداتیو کلینیک برگشت. استرس اکسیداتیو به عنوان اختلال در تعادل بین تولید اکسیژن فعال (رادیکال های آزاد) و دفاع آنتی اکسیدانی تعریف می شود. به عبارت دیگر، عدم تعادل بین تولید رادیکال های آزاد و توانایی بدن برای مقابله یا سم زدایی با اثرات مضر از طریق خنثی سازی توسط آنتی اکسیدان ها است. استرس اکسیداتیو منجر به بسیاری از شرایط پاتوفیزیولوژیکی در بدن می شود. اینها عبارتند از بیماری های تخریب کننده عصبی، به عنوان مثال، بیماری پارکینسون، بیماری آلزایمر، جهش های ژنی، سرطان ها، سندرم خستگی مزمن، سندرم X شکننده، اختلالات قلب و عروق خونی، تصلب شرایین، نارسایی قلبی، حمله قلبی، و بیماری های التهابی. اکسیداسیون تحت شرایط مختلفی اتفاق می افتد:
سلول ها برای تولید انرژی از گلوکز استفاده می کنند
سیستم ایمنی مبارزه با باکتری ها و ایجاد التهاب است
بدن دفع آلودگی ها، آفت کش ها و سیگار را دود می کند
میلیون ها فرآیند در بدن ما در هر زمان خاصی رخ می دهد که می تواند باعث اکسیداسیون شود. در اینجا چند نشانه وجود دارد:
خستگی
از دست دادن حافظه و یا مه
درد عضلانی و یا مفصلی
چین و چروک همراه موی خاکستری
کاهش بینایی
سردرد و حساسیت به سر و صدا
حساسیت به عفونت
انتخاب غذاهای ارگانیک و دوری از سموم در محیط شما تفاوت بزرگی ایجاد می کند. این امر در کنار کاهش استرس، می تواند در کاهش اکسیداسیون مفید باشد.
حتی بعد از شش ساعت خواب بیشتر از خواب بیدار می شوید؟
تحت فشار زیاد؟
اگر شما در حال تجربه هر یک از این شرایط هستید ، ممکن است به دلیل میزان ملاتونین و کورتیزول شما در بدن و ریتم شبانه روزی شما تأثیر بگذارد.
در سرتاسر جهان ، میلیون ها انسان در خوابیدن مشکل دارند. در ایالات متحده ، تقریباً وجود دارد 50-70 میلیون نفر که از کیفیت پایین خواب برخوردار نیستند وقتی فردی کمتر از هشت ساعت خوابیده باشد ، خسته می شود و مشکلات زیادی برای آنها بوجود می آید ، خصوصاً اگر زندگی آنها زیاد باشد. با داشتن سبک زندگی پرحرارت و خواب ناچیز می تواند باعث شود بدن برای انجام هر کار انرژی کمی داشته باشد ، هورمون استرس کورتیزول بالا می رود و بیماری هایی مانند فشار خون بالا و دیابت می تواند مشکلاتی ایجاد کند که در صورت نبودن مزمن باشد. تحت درمان.
در غدد درون ریز عملکردی ، ملاتونین و کورتیزول هورمون هایی هستند که بدن به طور طبیعی تولید می کند. هورمون کورتیزول یا هورمون استرس به بدن کمک می کند تا در حالت "جنگ یا پرواز" قرار داشته باشد ، که می تواند چیز خوبی برای هر کسی باشد که در حال انجام یک پروژه یا مصاحبه شغلی است. اگرچه هنگامی که سطح هورمون کورتیزول زیاد است ، می تواند باعث شود عارضه هایی مانند التهاب ، استرس اکسیداتیو مزمن و فشار خون بالا ایجاد شود.
ریتم شبانه روزی Melatonin
این هورمون به همراه هورمون ملاتونین به بدن می گوید که چه موقع خواب است. با این وجود ، بعضی اوقات خواب بسیار سختی در خواب است و مصرف مکمل های ملاتونین در واقع می تواند بدن را آرام کند و در نتیجه فرد را به خواب بیاندازد. از آنجایی که غده کاج ، ملاتونین را از مغز تولید می کند ، همچنین می توان در چشم ، مغز استخوان و روده برای آرامش بدن و ایجاد شخص در خواب طبیعی یافت. مقداری مطالعات نشان می دهد که ریتم شبانه روزی غده کاج که تولید ملاتونین است. با انجام این کار ، تحقیقات نشان می دهد که مصرف ملاتونین می تواند:
یک: به افرادی که مشکل خوابیدن دارند القا کنید.
دو: بدن را از بیدار شدن طبیعی از ضربان ساز شبانه روزی مهار می کند.
سه: ساعتهای بیولوژیکی شبانه روزی را تغییر دهید تا فرد هنگام خواب زودتر بخوابد تا از مزایای کامل هشت ساعته خواب ، خواب بیشتری ببرد.
وقتی شخصی در یک کار 9 تا 5 کار می کند ، با بدن خود در حال افزایش است و بدن خود را بعد از یک روز سخت در حال آرامش استراحت می کند. مطالعات دریافتند هورمونهای ملاتونین و کورتیزول به تنظیم الگوی 24 ساعته عملکرد بدن و پاسخ فوق العاده کمک می کند. با چرخه تولید هورمون بدن ، اگر فرد در اواخر شب بیدار بماند یا در طول روز بخوابد ، می تواند مختل شود. وقتی این اتفاق بیفتد ، فرد می تواند اختلالات ایجاد کنندگی مانند نوسانات خلقی ، سرگیجه ، تحریک پذیر و افسرده داشته باشد و اختلالات متابولیکی داشته باشد. نه تنها این ، بلکه سیستم ایمنی بدن و سیستم غدد درون ریز آن نیز می تواند آسیب ببیند ، و باعث می شود بدن میزبان عفونت ها و بیماری ها باشد.
مطالعات بیشتری در مورد ریتم شبانه روزی در بدن صورت گرفته است مطالعات نشان می دهد چگونگی کار افرادی که در شیفت شب کار می کنند با تعداد زیادی از مشکلات بهداشتی نامطلوب که به سیستم قلبی عروقی و دستگاه گوارش و همچنین اختلال در سیستم متابولیک حمله می کنند ، ارتباط دارد. هرکسی که در شیفت شب کار کرده است مجبور است برنامه خواب خود را تغییر داده و با برنامه ریزی مجدد سریع در برنامه خواب / بیداری خود سازگار شود تا به سر کار برود و کار خود را انجام دهد. از آنجا که همه در یک برنامه تغییر کار می کنند ، می تواند استرس زا باشد و می تواند بر عملکرد بدن یک کارگر و همچنین تأثیرگذاری بر ترشح ملاتونین و کورتیزول تأثیر بگذارد.
راه هایی برای حمایت از کورتیزول و ملاتونین
با کمال تعجب ، روش هایی برای پایین آمدن سطح کورتیزول وجود دارد و مطمئن شوید که سطح ملاتونین به درستی کار می کند تا بدن بتواند عملکرد خود را نشان دهد. برای پایین آمدن سطح کورتیزول ، فرد باید تمرینات مراقبتی انجام دهد ، یک سرگرمی لذت بخش پیدا کند و از همه مهمتر ، تمرینات تنفس عمیق را امتحان کنید تا بدن از استرس ناخواسته آرام شود. با انجام تمرینات تنفس عمیق ، می تواند به بدن کمک کند تا هر تنشی را که فرد در آن قرار دارد ، آزاد کند و ماهیچه های بدن شروع به آرامش کنند و خون شروع به جریان می کند. با مقادیر ملاتونین ، آنها با ریتم شبانه روزی بدن همکاری می کنند و مطمئن می شوند بدن چه موقع از خواب بیدار ، خواب و غذا می داند. هورمون ملاتونین همچنین می تواند به تنظیم دمای بدن کمک کند ، فشار خونو سطح هورمون برای اطمینان از عملکرد صحیح آن. هنگامی که سطح بالایی از این سیستم ها وجود دارد ، می تواند باعث شود بدن به بیماری های مزمن مبتلا شود و در این فرآیند به بدن آسیب برساند.
تحقیقات نشان میدهد هورمونهای ملاتونین می توانند به گیرنده های عصبی در بدن متصل شوند ، بنابراین باعث آرامش می شوند. از آنجایی که ملاتونین به گیرنده های عصبی متصل می شود ، همچنین می تواند باعث کاهش فعالیت عصبی و سطح دوپامین شود تا چشمان سنگین شود ، بنابراین فرد را به خواب می برد.
نتیجه
با این که بدن قادر است به طور طبیعی میزان ملاتونین و کورتیزول را تولید کند تا مطمئن شود که بدن در طول روز دچار فشار بیش از حد نمی شود. از آنجا که ملاتونین با ریتم شبانه روزی بدن شریک است ، بدن می داند چه زمانی بیدار شود و بخوابد. از آنجایی که همه افراد دارای یک برنامه گیج کننده هستند ، لازم است زمان و استراحت و برنامه سالم خواب را انجام دهید تا بدن سالم و کارآمد باشد. مقداری محصولات برای اطمینان از عملکرد صحیح سیستم غدد درون ریز و حمایت از غدد فوق کلیوی و متابولیسم قند در اینجا هستیم.
دامنه اطلاعات ما فقط به مباحث مربوط به کایروپراکتیک ، عضلانی و اسکلتی و سلامت اعصاب یا مقالات ، مباحث و مباحث پزشکی کاربردی محدود می شود. ما از پروتکل های بهداشتی عملکردی برای درمان آسیب ها یا اختلالات سیستم اسکلتی عضلانی استفاده می کنیم. دفتر ما تلاش منطقی را برای ارائه استنادهای حمایتی انجام داده است و مطالعه یا مطالعات تحقیقاتی مربوط به پست های ما را شناسایی کرده است. ما همچنین در صورت درخواست نسخه هایی از مطالعات تحقیقاتی پشتیبانی را در اختیار هیئت مدیره یا عموم قرار می دهیم. برای بحث بیشتر در مورد موضوع فوق ، لطفاً از دکتر الکس جیمنز سؤال کنید یا با ما در تماس باشید 915-850-0900.
منابع:
Cajochen، C، و همکاران. نقش ملاتونین در تنظیم ریتم شبانه روزی و خواب انسان. مجله علوم نورولوژي، کتابخانه ملی پزشکی ایالات متحده ، آوریل 2003 ، www.ncbi.nlm.nih.gov/pubmed/12622846.
جیمز، فرانسین او و همکاران. ریتمهای شبانهروزی ملاتونین، کورتیزول و بیان ژن ساعت در حین کار شبیهسازی شده در شیفت شب. خواب، انجمنهای خواب حرفه ای مرتبط ، LLC ، نوامبر 2007 ، www.ncbi.nlm.nih.gov/pmc/articles/PMC2082093/.
Monteleone، P، و همکاران. "رابطه زمانی بین پاسخ ملاتونین و کورتیزول به استرس فیزیکی شبانه در انسان". Psychoneuroendocrinology به، کتابخانه ملی پزشکی ایالات متحده ، 1992 ، www.ncbi.nlm.nih.gov/pubmed/1609019.
رامان، رایان. "چگونه ملاتونین می تواند به شما کمک کند تا بخوابید و احساس بهتری داشته باشید." Healthline، Healthline Media ، 3 سپتامبر 2017 ، www.healthline.com/nutrition/melatonin-and-sleep.
زمانیان، زهرا و همکاران. "بررسی تغییرات ریتم شبانه روزی کورتیزول و ملاتونین در حراست دانشگاه علوم پزشکی شیراز". مجله بین المللی پزشکی پیشگیری، انتشارات Med Knowledge & Media Pvt Ltd ، ژوئیه 2013 ، www.ncbi.nlm.nih.gov/pmc/articles/PMC3775223/.
با اطلاع رسانی به افراد در مورد چگونگی ارائه دانشگاه ملی علوم بهداشت دانش برای نسل های آینده که می خواهند در جهان تغییری ایجاد کنند ، فراهم می کند. دانشگاه طیف گسترده ای از حرفه های پزشکی را برای پزشکی کاربردی و یکپارچه ارائه می دهد.
تا به حال فکر کرده اید که چرا از یک روز طولانی احساس تنبلی می کنید؟ یا وقتی چیزی بد میخورید یا در غذای مورد علاقهتان زیادهروی میکنید، احساس ناراحتی میکنید؟ آیا ممکن است روده شما نشانه هایی از استرس و ناراحتی را به دلیل عادات خاصی که ممکن است با آنها روبرو شوید و حتی در مورد آن نمی دانستید نشان دهد؟
در مقاله قبلی ما در مورد آن صحبت کردیم شش نوع غذا که روده ما باید سالم باشد. از روده ما شامل تریلیون میکروبیوم ، چه خوب و چه بد ، این میکروبیوم ها نقش مهمی در سلامت کلی ما دارند. میکروبیوم سالم ما را بهبود می بخشد سلامت روده, سلامت قلب, سلامت مغز, وزن ما را کنترل می کند و قند خون ما را تنظیم می کند. باکتری های خوب موجود در روده ، باکتری ها از سیستم هضم خوبی برای ما بهره مند می شوند و باکتری های مضر را از بین می برند. اما برخی از شیوه های زندگی و رژیم های غذایی در واقع می توانند باکتری های بد را افزایش داده و باکتری های خوب و سلامت کلی را کاهش دهند.
در اینجا پنج انتخاب سبک زندگی شگفت آور که به روده شما آسیب می رساند ، آورده شده است:
خوردن طیف گسترده ای از غذاها
روده ما نقش مهمی در سلامت کلی ما دارد. وقتی غذاهای کامل بخوریم ، روده ما شادتر است. ما انرژی بیشتری برای انجام هر وظیفه ای که بر ما وارد شده و در حال انجام آن هستیم ، داریم مواد مغذی برای گیاهان روده ما. با این حال ، در طول چند دهه گذشته ، ما به دلیل فشارهای اقتصادی افزایش تولید محصولات غذایی ، بیشتر به غذاهای فرآوری شده متمایل شده ایم. FOA بیان کرد که 75 درصد غذای جهان تنها از 12 گونه گیاهی و پنج گونه جانوری تولید می شود و این برای فلور روده ما بسیار بد است.
در اینجا در کلینیک پزشکی و عمل جراحی Injury ، ما بیماران خود را در مورد اهمیت خوردن غذاهای مغذی و کامل برای ارتقا promote نه تنها روده سالم بلکه ذهن سالم آگاه می کنیم. وقتی بدن به a معرفی می شود طیف گسترده ای از غذاهای کامل (با داشتن فیبر زیاد) ، روده ما شروع به ترمیم آسیب مواد غذایی فرآوری شده که ممکن است در داخل کشور مصرف کرده باشیم ، می کند.
با این حال ، هنگامی که شما از prebiotics به رژیم خود چشم پوشی می کنید ، شما هستید به سلامتی گوارش شما آسیب می رساند. بدون پروبیوتیک ، سیستم گوارشی ما رشد و تنوع گیاهان روده ما را کند می کند. بنابراین به منظور داشتن میکروبیوم سالم توسعه ، شما باید غذاهای پر از فیبر قابل هضم و غیر قابل هضم را در رژیم غذایی خود بگنجانید. برخی غذاهای موجود در این دسته عبارتند از: جو ، آجیل ، پیاز ، سیر ، تره فرنگی ، مارچوبه ، موز ، گلابی ، نخود و لوبیا.
چسباندن رژیم غذایی با فیبر بالا ممکن است چالش برانگیز باشد ، اما گزینه مصرف مکمل های پربیوتیک نیز وجود دارد. اگر حساسیت به مواد غذایی یا حساسیت غذایی به هر نوع غذای غنی شده با فیبر دارید ، مصرف کنید مکمل های پری بیوتیک در واقع می تواند به رشد Bifidobacterium و Faecalibacterium در روده شما کمک کند و بدون ناراحتی برای سلامتی شما مفید باشد.
مصرف بیش از حد الکل
هر بزرگسالی هر چند وقت یکبار از الکل لذت می برد. بله، این یکی از آن نوشیدنی هایی است که به شما کمک می کند بعد از یک روز طولانی کمی آرام شوید، با این حال، مصرف بیش از حد آن می تواند منجر به سوء مصرف الکل و اعتیاد شود. بنابراین، آیا می دانستید که مصرف این مقدار الکل برای بدن مضر است قلب ، کبد و مغز شما؛ بنابراین به سلامتی روده شما آسیب می رسانید و به شما دیس بیضه می دهد؟
یک مطالعه بیان کرد که افراد الکلی مبتلا به دیس بیوز دارای میانگین فراوانی کمتری از باکتریها و فراوانی بالایی از پروتئوباکتریها هستند. آنهایی که الکلی نبودند تحت تأثیر این مطالعه قرار نگرفتند.
با این حال؛ خبرهای خوبی درباره محدود کردن خود به الکلیسم وجود دارد و این می تواند برای باکتریهای روده شما مفید باشد. اگر معتدل شراب قرمز را با مسئولیت پذیری مصرف می کنید ، پلی در شراب می تواند به نفع فلور روده شما کمک کند. بنابراین ، یک مرتبه از یک لیوان شراب به عنوان یک درمان کوچک که نباید آن را به صورت اعطا پذیرفت لذت ببرید.
خواب ناکافی
در یکی از مقاله های قبلی ، در مورد چگونگی دستیابی به a صحبت کردیم شب بخیر خواب از طریق گیاهان هنگامی که در طول زندگی گیج کننده خود کمی خوابیدیم ، از طریق مشکلات مختلف سلامتی ، از جمله ، ما را تحت تأثیر قرار می دهد بیماری قلبی و چاقی. در یک مطالعه 2016محققان بعد از گذشت دو روز ، اثر کمبود خواب کوتاه مدت بر میکروبیوتای روده را کشف کردند.
وقتی بدن ما 8 ساعت خواب توصیه شده را دریافت نمی کند، روده ما آسیب زیادی می بیند زیرا ما احساس سستی و خستگی می کنیم. بنابراین، برای اطمینان از اینکه ما میکروبیوم روده مراقبت می شود، توصیه می کنیم حداقل 30 دقیقه قبل از اینکه برای استراحت شبانه آماده شوید، دستگاه های الکترونیکی خود را خاموش کنید. تمام چراغ ها را خاموش کنید و حداقل دو ساعت قبل از خواب هیچ مایعی ننوشید، چشمان خود را ببندید و در حالت مراقبه نفس عمیق بکشید و در حالی که به سمت شهر خواب می روید استراحت کنید.
ورزش نامناسب
از طریق سبک زندگی سریع و مشاغل پر استرس ما، یافتن زمانی برای ورزش دشوار است. اما وقتی واقعاً زمانی برای ورزش پیدا می کنیم، نه تنها ذهنمان احساس خوبی می کند. اما بدن و روده ما نیز احساس خوبی دارند. با این حال، همیشه وقتی در یک برنامه ورزشی قرار می گیریم و مجبوریم به طور کلی از ورزش صرف نظر کنیم، همه چیز پیش می آید. این برای همه ما اتفاق میافتد و دشوار است که از جایی که زمانی که سعی کردیم ورزش کنیم ادامه دهیم.
وقتی حداقل چند بار در هفته ورزش نمی کنیم، بدن ما با افزایش وزن، آسیب زیادی به ما وارد می کند. استرس خیلی زیاد است، و ما یک شانس بالاتر از ابتلا به یک بیماری مزمن وقتی این اتفاق می افتد فلور روده ما یک نقطه ضعف بزرگ است. در اینجا در کلینیک ، ما تلاش داریم تا بیماران خود را در مورد اهمیت ورزش آگاه سازیم و این نه تنها زندگی آنها را تغییر می دهد بلکه روحیه آنها را به کلی تغییر می دهد.
با این حال، فقط وارد یک برنامه تمرینی سخت نشوید که در آن به خود صدمه می زنید. با یک تمرین با شدت کم شروع کنید و در حین حرکت آن را تقویت کنید زیرا فلور روده شما به خاطر آن از شما تشکر خواهد کرد.
به عنوان یک گفته نهایی ، ما در اینجا در Injury Medical می خواهیم شما را در مورد تغذیه و راه هایی برای کمک به بهبود بیماری های خود با این شگفتی های 5 آگاه کنیم. اما همچنین به شما آموزش اینکه چه چیزی ممکن است به روده شما آسیب برساند. با این شگفتی ها و تغییرات جزئی در زندگی روزمره ، روده شما از مسافت طولانی تشکر می کند.
منابع NCBI
طبق شواهد حاصل از یک مطالعه تحقیقاتی 2016، سیستم ایمنی روده برای پیشگیری از انواع بیماری ها اساسی است و اغلب ممکن است به اختلالات متابولیک کمک کند. با این حال، ممکن است در هنگام مشاهده التهاب سیستمیک در مقاومت به انسولین، به ارائه یک هدف درمانی نیز کمک کند. علاوه بر این، ایمنی اصلاح شده روده با تغییراتی در میکروبیوتای روده، عملکرد سد روده، سلول های ایمنی ساکن روده و مقاومت در برابر آنتی ژن هایی که وارد سیستم گوارشی یا GI می شوند مرتبط است. اگرچه قبلاً اعتقاد بر این بود که این امر خطر بیماریهای مری از جمله عفونتهای بیماریزا و التهاب مزمن را افزایش میدهد که در نهایت ممکن است منجر به مشکلات سلامت مزمن شود.
بدن کتون توسط کبد ایجاد می شود و هنگامی که گلوکز به راحتی در بدن انسان در دسترس نیست ، به عنوان منبع انرژی مورد استفاده قرار می گیرد. دو جسم اصلی کتون عبارتند از استو استات (AcAc) و 3-بتا-هیدروکسی بوتیرات (3HB) ، در حالی که استون سومین و کمترین جسم کتون است. کتونها همیشه در خون وجود دارند و در طی روزه داری و ورزش طولانی مدت سطح آنها افزایش می یابدKetogenesis فرآیند بیوشیمیایی است که توسط آن ارگانیزم بدن کتون را از طریق تجزیه اسید های چرب و اسیدهای آمینه کتوژن تولید می کند.
بدن کتون در اکثر موارد تولید می شود میتوکندری سلول های کبدی. کتوژنز زمانی اتفاق می افتد که سطح گلوکز پایین در خون وجود داشته باشد، به ویژه پس از اینکه دیگر فروشگاه های کربوهیدرات سلولی مانند گلیکوژن خسته شده اند. این مکانیسم همچنین می تواند زمانی رخ دهد که مقدار کافی انسولین وجود نداشته باشد. در نهایت تولید کتونهای بدن برای ایجاد انرژی در دسترس است که در بدن انسان به عنوان اسید چرب ذخیره می شود. کتوژنز در میتوکندری رخ می دهد که در آن به طور مستقل تنظیم می شود.
چکیده
متابولیسم بدن کتونی یک گره مرکزی در خانه شناسی فیزیولوژیک است. در این بررسی، ما بحث می کنیم که چگونه کتون ها نقش متابولیک ریز تنظیم کننده ای را ایفا می کنند که عملکرد ارگان و ارگانیزم را در انواع باقی مانده های مواد مغذی بهینه می کند و از التهاب و آسیب در سیستم های چندگانه محافظت می کند. به طور سنتی به عنوان زیر ساختارهای متابولیک تنها در محدودیت کربوهیدرات مورد توجه قرار گرفته است، مشاهدات اخیر، اهمیت بدن کتون را به عنوان متصديان متابوليسم و سيگنالينگ مهم در هنگام کربوهيدرات فراوان است. با تکمیل مجموعه ای از گزینه های درمان شناخته شده برای بیماری های سیستم عصبی، نقش های آینده ای برای بدن کتون در سرطان ایجاد شده است، به عنوان نقش محافظتی جذاب در قلب و کبد، باز کردن گزینه های درمانی در بیماری های مرتبط با چاقی و قلب و عروق. اختلاف نظر در متابولیسم کوانتوم و سیگنالینگ به منظور مقابله با داستانی کلاسیک با مشاهدات معاصر مورد بحث قرار گرفته است.
معرفی
اجسام کتون یک منبع سوخت متابولیکی جایگزین حیاتی برای تمام حوزه های زندگی ، یوکاریا ، باکتری ها و باستان است (Aneja et al.، 2002؛ Cahill GF Jr، 2006؛ Krishnakumar et al.، 2008). متابولیسم بدن کتون در انسان برای سوخت رسانی به مغز در دوره های اپیزودیک محرومیت از مواد مغذی استفاده شده است. اجسام کتون با مسیرهای متابولیکی حیاتی پستانداران مانند بتاکسیداسیون (FAO) ، چرخه اسید تریکاربوکسیلیک (TCA) ، گلوکونئوژنز ، لیپوژنز نو (DNL) و بیوسنتز استرول ها در هم تنیده شده اند. در پستانداران ، اجسام کتون عمدتا از استیل-CoA مشتق شده از فاو در کبد تولید می شوند و آنها برای اکسیداسیون ترمینال به بافتهای خارج کبدی منتقل می شوند. این فیزیولوژی سوخت جایگزینی را تأمین می کند که با دوره های نسبتاً کوتاه ناشتایی تقویت می شود ، که در دسترس بودن اسیدهای چرب را افزایش می دهد و میزان کربوهیدرات را کاهش می دهد (Cahill GF Jr، 2006؛ McGarry and Foster، 1980؛ Robinson and Williamson، 1980). اکسیداسیون بدن کتون به میزان قابل توجهی در متابولیسم انرژی پستانداران در بافتهای خارج کبدی در هزاران حالت فیزیولوژیکی از جمله روزه داری ، گرسنگی ، دوره نوزادی ، بعد از ورزش ، بارداری و پیروی از رژیم های کم کربوهیدرات نقش دارد. غلظت کل بدن کتون در گردش در انسان های بالغ سالم به طور معمول بین تقریباً 100 250 M دارای نوسانات شبانه روزی است ، پس از ورزش طولانی مدت یا 1 ساعت ناشتا به 24 میلی متر افزایش می یابد و می تواند در حالت های پاتولوژیک مانند کتواسیدوز دیابتی تا 20 میلی متر تجمع یابد ( Cahill GF Jr، 2006؛ Johnson et al.، 1969b؛ Koeslag et al.، 1980؛ Robinson and Williamson، 1980؛ Wildenhoff et al.، 1974). کبد انسان روزانه حداکثر 300 گرم بدن کتون تولید می کند (بالاس و فری ، 1989) ، که 5 تا 20٪ از کل انرژی مصرفی در حالت های تغذیه ، روزه و گرسنگی را به خود اختصاص می دهد (بالاس و همکاران ، 1978 ؛ کوکس و al. ، 2016).
در حال حاضر مطالعات اخیر، نقش مهمی برای بدن کتون در متابولیسم سلول های پستانداران، هوموتازیست و سیگنالینگ تحت انواع مختلف بیماری های فیزیولوژیکی و پاتولوژیک را برجسته می کند. به جز استفاده از سوخت های انرژی برای بافت های خارج از بافت مثل مغز، قلب و عضلات اسکلتی، بدن کتون نقش اساسی را به عنوان واسطه های سیگنالینگ، رانندگان تغییرات پس از ترجمه پروتئین (PTM) و مدولاتورهای التهاب و استرس اکسیداتیو ایفا می کند. در این بررسی، ما هر دو دیدگاه های کلاسیک و مدرن از نقش های پلئوتروپیک بدن های کتون و متابولیسم آنها را ارائه می دهیم.
بررسی کلی متابولیسم بدن کتون
میزان کتوژنز کبدی توسط یک سری تنظیم شده از تغییرات فیزیولوژیکی و بیوشیمیایی چربی کنترل می شود. تنظیم کننده های اولیه شامل لیپولیز اسیدهای چرب از تریآسیل گلیسرول ها ، انتقال به داخل و غشا plas پلاسمای سلولهای کبدی ، انتقال به میتوکندری از طریق کارنیتین پالمیتوئیل ترانسفراز 1 (CPT1) ، مارپیچ اکسیداسیون؟ ، فعالیت چرخه TCA و غلظت های میانی ، پتانسیل اکسایش و تنظیم کننده های هورمونی است. از این فرآیندها ، عمدتا گلوکاگون و انسولین [بررسی شده در (Arias و همکاران ، 1995 ؛ Ayte و همکاران ، 1993 ؛ Ehara و همکاران ، 2015 ؛ Ferre و همکاران ، 1983 ؛ Kahn و همکاران ، 2005 ؛ McGarry و Foster ، 1980 ؛ ویلیامسون و دیگران ، 1969)]. به طور کلاسیک کتوژنز به عنوان یک مسیر سرریز مشاهده می شود ، که در آن استیل- CoA حاصل از اکسیداسیون بیش از فعالیت سنتاز سیترات و / یا در دسترس بودن اکسالواستات برای میعان تشکیل سیترات است. واسطه های سه کربنی فعالیت ضد کتوژنیک از خود نشان می دهند ، احتمالاً به دلیل توانایی آنها در گسترش استخر اگزالو استات برای مصرف استیل-CoA ، اما غلظت استیل-CoA کبدی به تنهایی میزان کتوژنیک را تعیین نمی کند (فاستر ، 1967 ؛ راوات و مناهان ، 1975 ؛ ویلیامسون و همکاران ، 1969). تنظیم کتوژنز توسط حوادث هورمونی ، رونویسی و پس از ترجمه با هم این عقیده را پشتیبانی می کند که مکانیسم های مولکولی سرعت کتوژنیک دقیق را کاملاً درک نشده باقی می مانند (به مقررات HMGCS2 و SCOT / OXCT1 مراجعه کنید).
کتوژنز در درجه اول در ماتریس میتوکندری کبدی با نرخ متناسب با اکسیداسیون کل چربی رخ می دهد. پس از انتقال زنجیره های آسیل از طریق غشاهای میتوکندری و اکسیداسیون β ، ایزوفرم میتوکندری 3-هیدروکسی متیل گلوتاریل-کوآ سنتاز (HMGCS2) سرنوشت متعهد شدن متراکم استواستیل CoA (AcAc-CoA) و استیل CoM CoA را تولید می کند (شکل 1 الف) HMG-CoA لیاز (HMGCL) باعث تجزیه HMG-CoA برای آزادسازی استیل-CoA و استواستات (AcAc) می شود و دومی توسط d-؟ OHB دهیدروژناز وابسته به فسفاتیدیل کولین به d -؟ - هیدروکسی بوتیرات (d-؟ OHB) کاهش می یابد. BDH1) در یک واکنش نزدیک به تعادل NAD + / NADH بهم پیوسته (Bock and Fleischer، 1975؛ LEHNINGER et al.، 1960). ثابت بودن تعادل BDH1 به تولید d-؟ OHB علاقه مند است ، اما نسبت اجسام کتون AcAc / d-؟ OHB مستقیماً با نسبت NAD + / NADH میتوکندری متناسب است و بنابراین فعالیت اکسیدوروکتاز BDH1 پتانسیل اکسایش میتوکندری را تعدیل می کند (Krebs و همکاران ، 1969 ؛ ویلیامسون و دیگران ، 1967). AcAc همچنین می تواند به طور خودبه خود از استون (پدرسن ، 1929) ، منبع بوی شیرین در انسانهایی که دچار کتواسیدوز هستند ، دکربوکسیله شود (به عنوان مثال ، اجسام کتون سرم کلی> 7 میلی متر ~ میلی متر ؛ AcAc pKa 3.6 ،؟ OHB pKa 4.7). مکانیسم هایی که از طریق آنها اجسام کتون از طریق غشای داخلی میتوکندری منتقل می شود شناخته شده نیست ، اما AcAc / d-؟ OHB از طریق حمل کننده های مونوکربوکسیلات از سلول ها آزاد می شود (در پستانداران ، MCT 1 و 2 ، همچنین به عنوان حامل املاح 16A اعضای خانواده 1 و 7 شناخته می شود) 2011) و برای گردش در بافتهای خارج کبدی برای اکسیداسیون ترمینال منتقل می شود (Cotter et al.، 2012؛ Halestrap and Wilson، 2012؛ Halestrap، 2012؛ Hugo et al.، 1940). غلظت اجسام کتون در گردش بیشتر از آن است که در بافتهای خارج کبدی وجود دارد (هریسون و لانگ ، 1) که نشان می دهد اجسام کتون به یک شیب غلظت منتقل می شوند. جهش های از دست دادن عملکرد در MCTXNUMX با دوره های خود به خودی کتواسیدوز همراه است ، که نقش مهمی در واردات بدن کتون دارد.
به استثنای انحراف بالقوه اجسام کتون به سرنوشتهای غیر اکسیداتیو (به سرنوشت متابولیکی غیر اکسیداتیو بدن کتون نگاه کنید) ، سلولهای کبدی توانایی متابولیسم اجسام کتونی تولیدی را ندارند. اجسام کتونی سنتز شده توسط کبد از طریق کبد (i) در میتوکندری بافتهای خارج کبدی به استیل-CoA کاتابولیزه می شوند ، که برای اکسیداسیون ترمینال در دسترس چرخه TCA است (شکل 1 A) ، (ii) به مسیرهای لیپوژنز یا سنتز استرول هدایت می شوند ( شکل 1B) ، یا (III) از طریق ادرار دفع می شود. به عنوان یک سوخت جایگزین انرژی ، اجسام کتون در قلب ، عضله اسکلتی و مغز اکسید می شوند (بالاسه و فری ، 1989 ؛ بنتورکیا و دیگران ، 2009 ؛ اوون و همکاران ، 1967 ؛ ریچارد و همکاران ، 1974 ؛ سلطان ، 1988 ) میتوکندری خارج کبدی BDH1 اولین واکنش اکسیداسیون؟ OHB را کاتالیز می کند و آن را به AcAc تبدیل می کند (LEHNINGER et al.، 1960؛ Sandermann et al.، 1986). یک d-؟ OHB- دهیدروژناز سیتوپلاسمی (BDH2) فقط با توالی 20٪ BDH1 دارای کیلومتر زیاد برای اجسام کتون است و همچنین در هموستاز آهن نقش دارد (داولوری و همکاران ، 2016 ؛ گوو و همکاران ، 2006) . در ماتریس میتوکندری خارج کبدی ، AcAc از طریق تبادل یک بخش CoA از سوکسینیل-CoA در یک واکنش کاتالیز شده توسط یک ترانسفراز CoA پستانداران منحصر به فرد ، سوکسینیل-CoA: ترانسفراز 3-اکسواسید-CoA (SCOT ، CoA ترانسفراز) به AcAc-CoA فعال می شود. رمزگذاری شده توسط OXCT1) ، از طریق یک واکنش تعادل نزدیک. انرژی آزاد آزاد شده توسط هیدرولیز AcAc-CoA بیشتر از سوکسینیل CoA است ، به نفع تشکیل AcAc است. بنابراین شار اکسیداتیو بدن کتون به دلیل عمل جرم اتفاق می افتد: یک منبع فراوان AcAc و مصرف سریع استیل-CoA از طریق سیترات سنتاز باعث ایجاد AcAc-CoA (+ سوکسینات) توسط SCOT می شود. قابل ذکر است ، بر خلاف گلوکز (هگزوکیناز) و اسیدهای چرب (آسیل-CoA سنتاز) ، فعال شدن اجسام کتون (SCOT) به شکل اکسید شونده نیازی به سرمایه گذاری ATP ندارد. یک واکنش تیولاز AcAc-CoA برگشت پذیر [توسط هر یک از چهار تیولاز میتوکندری کدگذاری شده توسط ACAA2 (رمزگذاری آنزیمی معروف به T1 یا CT) ، ACAT1 (رمزگذار T2) ، HADHA یا HADHB] کاتالیز می شود دو مولکول استیل-CoA تولید می کند ، که وارد چرخه TCA می شوند (Hersh and Jencks، 1967؛ Stern et al.، 1956؛ Williamson et al.، 1971). در طی حالت های کتوسی (یعنی کل کتونهای سرم> 500 میلی متر) ، بدن کتونها به میزان قابل توجهی در مصرف انرژی نقش دارند و در بافتها به سرعت مورد استفاده قرار می گیرند تا زمان جذب یا اشباع اکسیداسیون (Balasse et al.، 1978؛ Balasse and Fery، 1989) ؛ ادموند و همکاران ، 1987). کسر بسیار کمی از اجسام کتون مشتق از کبد را می توان به راحتی در ادرار اندازه گیری کرد ، و میزان استفاده و جذب مجدد توسط کلیه متناسب با غلظت گردش خون است (گلدشتاین ، 1987 ؛ رابینسون و ویلیامسون ، 1980). در طی حالتهای بسیار کتوتیک (> 1 میلی مولار در پلاسما) ، کتونوریا به عنوان گزارشگر کمی کمی کتوز عمل می کند ، اگرچه بیشتر آزمایشات بالینی بدن کتون ادرار AcAc را تشخیص می دهند اما نه OHB (Klocker و همکاران ، 2013)
بسترهای کتوژنیک و تأثیر آنها بر متابولیسم هپاتوستیک
بسترهای کتوژنیک شامل اسیدهای چرب و اسیدهای آمینه هستند (شکل 1B). کاتابولیسم اسیدهای آمینه، به خصوص لوسین، در حدود 4٪ از اجسام کتون در حالت پس از جذب (Thomas et al.، 1982) تولید می شود. به همین دلیل، استاتیک CoA برای ساخت کتون ها به طور عمده از اسیدهای چرب ساخته می شود، زیرا در حالت های کاهش کربوهیدرات، پیوروت به چرخه TCA کبدی وارد می شود، به ویژه از طریق آناپلروز، یعنی کربوکسیلسیون وابسته به ATP به اگزالاکتیت (OAA) یا مالات (MAL)، و نه decarboxylation اکسیداتیو به acetyl-CoA (Jeoung و همکاران، 2012؛ Magnusson و همکاران، 1991؛ Merritt و همکاران، 2011). در کبد، گلوکز و پیوواست نقش مهمی در کتوژنز دارند، حتی وقتی که decarboxylation پیروات به استیل کولا بیشترین مقدار را دارد (Jeoung و همکاران، 2012).
استیل کوا چندین نقش متمایز کننده متابولیسم واسطه های کبدی را فراتر از تولید ATP از طریق اکسیداسیون ترمینال شامل می کند (همچنین ادغام متابولیسم بدن کتون، اصلاح بعد از ترجمه و فیزیولوژی سلولی). Acetyl-CoA آلوستریک (I) پیروات کربوکسیلاز (PC) را فعال می کند، در نتیجه مکانیسم کنترل متابولیک را فعال می کند که ورود آناپلیروئید متابولیت ها را به چرخه TCA افزایش می دهد (Owen و همکاران، 2002، Scrutton و Utter، 1967) و (ii) پیرووات دهیدروژناز کیناز، که فسفوریلات و پریوروآدروژناز (PDH) را مهار می کند (کوپر و همکاران، 1975)، در نتیجه موجب افزایش جریان پیروات در چرخه TCA از طریق آناپلروز می شود. علاوه بر این، استیل کلاسی سیتوپلاسمی که استحکام آن توسط مکانیسم هایی که استیل کولیت میتوکندری را به متابولیت های قابل حمل تبدیل می کند، اکسیداسیون اسید های چرب را مهار می کند: کراتوکولاز استیل کروم (ACC) کاتالیز کننده تبدیل استیل CoA به ملونیل-CoA، سوبسترا لیپوژنیک و مهار کننده آلوستریک CPT1 میتوکندری [در بررسی (Kahn و همکاران، 2005، McGarry و Foster، 1980)] بررسی شده است. بنابراین، استحکام میتوکندری استیل CoA هر دو تنظیم شده و توسط مسیر عبور از کتوژنز تنظیم شده است، که ارکان اصلی جنبه های متابولیسم واسطه های کبدی است.
غیر متخلخل متابولیک بدن کتون
سرنوشت غالب کتونهای مشتق شده از کبد، وابسته به SCOT اکسیداسیون فوق العاده ای است. با این حال، AcAc می تواند از میتوکندری صادر شود و از طریق تبدیل به AcAc-CoA با استفاده از واکنش وابسته به ATP کاتالیز شده توسط سیتوپلاسمی acetoacetyl-CoA synthetase (AACS، شکل 1B) در مسیرهای آنابولیک مورد استفاده قرار می گیرد. این مسیر در حین رشد مغز و شیردهی شیردهی (Morris، 2005، Robinson و Williamson، 1978، Ohgami و همکاران، 2003) فعال است. AACS نیز به شدت در بافت چربی و استئوکلاست فعال شده است (Aguilo و همکاران، 2010، Yamasaki و همکاران، 2016). AcAc-CoA سیتوپلاسمی می تواند به واسطه سیتوزول HMGCS1 به سمت بیوسنتز استرول یا توسط هر دو تيولاز سيتوپلاسمی به استيل CoA (ACAA1 و ACAT2)، کربوکسیل شده به مالونيل-CoA، و به سنتز اسيدهاي چرب (Bergstrom et al.، 1984؛ ادموند، 1974؛ Endemann و همکاران، 1982؛ ژلن و همکاران، 1983؛ وببر و ادموند، 1977).
در حالی که اهمیت فیزیولوژیکی هنوز مشخص نشده است ، کتونها حتی در کبد می توانند به عنوان بسترهای آنابولیک عمل کنند. در زمینه های آزمایشگاهی مصنوعی ، AcAc می تواند به اندازه نیمی از لیپید تازه سنتز شده و تا 75٪ کلسترول جدید سنتز شده کمک کند (Endemann et al.، 1982؛ Geelen et al.، 1983؛ Freed et al.، 1988). از آنجا که AcAc از اکسیداسیون ناقص چربی کبدی گرفته شده است ، توانایی AcAc برای کمک به لیپوژنز در داخل بدن به معنای دوچرخه سواری بیهوده کبدی است ، جایی که می توان از کتونهای مشتق شده از چربی برای تولید چربی استفاده کرد ، مفهومی که اهمیت فیزیولوژیکی آن نیاز به اعتبار آزمایشی دارد ، اما می تواند مفید باشد نقش های سازگار یا ناسازگار (Solinas و همکاران ، 2015). AcAc مشتاقانه کلستروژنز را تهیه می کند ، با AACS Km-AcAc کم (50 MM) که حتی در حالت تغذیه فعال AcAc را ترجیح می دهد (Bergstrom و همکاران ، 1984). نقش پویای متابولیسم کتون سیتوپلاسمی در سلولهای عصبی جنینی موش اولیه و سلولهای چربی مشتق شده از 3T3-L1 پیشنهاد شده است ، زیرا AACS باعث کاهش اختلال در تمایز هر نوع سلول می شود (Hasegawa et al.، 2012a؛ Hasegawa et al.، 2012b). از بین بردن AACS در موش های داخل بدن باعث کاهش کلسترول سرم می شود (Hasegawa و همکاران ، 2012c). SREBP-2 ، تنظیم کننده اصلی رونویسی بیوسنتز کلسترول ، و گیرنده فعال کننده تکثیر پروکسی زوم (PPAR) -؟ فعال کننده های رونویسی AACS هستند و رونویسی آن را در طی رشد نوریت و در کبد تنظیم می کنند (Aguilo et al.، 2010؛ Hasegawa et al.، 2012c). در مجموع ، متابولیسم بدن کتون سیتوپلاسمی ممکن است در شرایط خاص یا تاریخ طبیعی بیماری مهم باشد ، اما برای دفع اجسام کتون مشتق شده از کبد کافی نیست ، زیرا هیپرکتونمی عظیم در تنظیم اختلال انتخابی سرنوشت اکسیداتیو اولیه از طریق از دست دادن جهش های عملکردی رخ می دهد به SCOT (بری و همکاران ، 2001 ؛ کوتر و همکاران ، 2011).
مقررات HMGCS2 و SCOT / OXCT1
واکنش یک میتوکندریایی از ژن کدگذاری HMGCS سیتوزول در اوایل تکامل مهره ها به علت نیاز به حمایت از کتوژنز کبدی در گونه های با نسبت مغز نسبت به وزن بدن (Boukaftane و همکاران، 1994، Cunnane و Crawford، 2003) رخ داده است. جهش های HMGCS2 از دست دادن طبیعت در انسان منجر به بروز هیپوگلیسمی هیپوکتوئیک می شوند (Pitt et al.، 2015؛ Thompson et al.، 1997). بیان قوی HMGCS2 به هپاتوسیت ها و اپیتلیوم کولون محدود می شود و بیان و فعالیت آنزیمی از طریق مکانیزم های مختلف (Mascaro و همکاران، 1995، مک گرری و فاستر، 1980، رابینسون و ویلیامسون، 1980) هماهنگ می شوند. در حالی که محدوده کامل حالت های فیزیولوژیکی که HMGCS2 را تحت تاثیر قرار می دهند، نیاز به توضیح بیشتری دارد، بیان و / یا فعالیت آن در طی دوره اولیه پس از تولد، پیری، دیابت، گرسنگی یا مصرف رژیم کتوژنیک (Balasse and Fery، 1989، Cahill GF Jr، 2006 ؛ Girard و همکاران، 1992؛ هگارد، 1999؛ ساتاپتی و همکاران، 2012؛ Sengupta و همکاران، 2010). در جنین ، متیلاسیون منطقه 5 طرفین ژن Hmgcs2 با رونویسی آن رابطه معکوس دارد و پس از تولد تا حدی معکوس می شود (آریاس و همکاران ، 1995 ؛ آیته و دیگران ، 1993 ؛ Ehara و همکاران ، 2015 ؛ Ferre و همکاران . ، 1983). به طور مشابه، Bdh1 کبدی دارای الگوی بیان تکاملی است که از زمان تولد تا از بین بردن آن افزایش می یابد و همچنین توسط رژیم کتوژنیک در یک فاکتور رشد فیبروبلاست (FGF) -21 بوجود می آید (Badman et al.، 2007، Zhang et al.، 1989 ) کتوژنز در پستانداران به شدت به انسولین و گلوکاگون واکنش نشان می دهد که به ترتیب سرکوب و تحریک می شوند (McGarry and Foster، 1977). انسولین لیپولیز بافت چربی را مهار می کند، بنابراین کتوژنز بستر خود را محروم می کند، در حالی که گلوکاگون باعث افزایش قدرت کتوژنیک از طریق اثر مستقیم بر کبد می شود (هگارد، 1999). Hmgcs2 رونویسی است forkhead FOXA2 عامل رونویسی، که از طریق انسولین فسفاتیدیل-3 کیناز / AKT مهار تحریک می شوند، و توسط گلوکاگون اردوگاه-p300 ناشی از زنگ (آریاس و همکاران، 1995؛ Hegardt، 1999؛ م و همکاران ، 1990؛ Thumelin و همکاران، 1993؛ فون Meyen و همکاران، 2013؛ Wolfrum و همکاران، 2004؛ Wolfrum و همکاران، 2003). PPAR؟ (رودریگز و همکاران ، 1994) همراه با هدف خود ، FGF21 (بادمن و همکاران ، 2007) همچنین باعث تحریک رونویسی Hmgcs2 در کبد در هنگام گرسنگی یا تجویز رژیم کتوژنیک می شود (بادمن و دیگران ، 2007 ؛ ایناگاکی و همکاران ، 2007 ) القای PPAR؟ ممکن است قبل از انتقال از فیزیولوژی جنین به نوزاد اتفاق بیفتد ، در حالی که فعال سازی FGF21 ممکن است در اوایل دوره نوزادی از طریق مهار OHB واسطه دیاستیلاز هیستون (HDAC) -3 (Rando و همکاران ، 2016) مورد علاقه قرار گیرد. مهار وابسته به PPAR mTORC1 (هدف پستانداران کمپلکس رپامایسین 1)؟ فعالیت رونویسی نیز تنظیم کننده کلیدی بیان ژن Hmgcs2 است (Sengupta و همکاران ، 2010) ، و کبد PER2 ، یک نوسان دهنده اصلی شبانه روزی ، به طور غیر مستقیم بیان Hmgcs2 را تنظیم می کند (Chavan و همکاران ، 2016). مشاهدات اخیر نشان می دهد که اینترلوکین -6 ناشی از تومور خارج کبدی باعث اختلال در کتوژنز از طریق PPAR می شود؟ سرکوب (Flint و همکاران ، 2016).
فعالیت آنزیم HMGCS2 از طریق چندین PTM تنظیم می شود. فسفوریلاسیون سریین HMGCS2 فعالیت خود را در شرایط آزمایشگاهی افزایش داد (Grimsrud et al.، 2012). . Hegardt، 2؛ لاو و تابز، 1995؛ م و همکاران، 1999؛ Rardin و همکاران، 1985؛ فعالیت HMGCS1990 است allosterically توسط سوکسینیل کوآ و لیزین succinylation باقی مانده (آریاس و همکاران، 2013 مهار رید و همکاران،. 1975؛ Thumelin و همکاران، 1993). سوسینیلینگ باقی مانده های لیزین HMGCS2، HMGCL و BDH1 در میتوکندری های کبدی، اهداف وابسته به DAD + وابسته به دیازیلاز سورونتین 5 (SIRT5) (Rardin و همکاران، 2013) است. فعالیت HMGCS2 نیز توسط de-acetylation لیزین SIRT3 افزایش یافته است و ممکن است که تداخل بین استیلیت و سوکسینسیون تنظیم فعالیت HMGCS2 (Rardin و همکاران، 2013، Shimazu و همکاران، 2013). علیرغم توانایی این PTM ها برای تنظیم HMGCS2 Km و Vmax، نوسانات این PTM ها هنوز با دقت نقشه بندی نشده اند و به عنوان رانندگان مکانیکی کتوژنز in vivo تایید نشده اند.
SCOT در تمام سلول های پستاندارانی که دارای میتوکندری هستند به جز hepatocytes ها بیان می شود. اهمیت فعالیت SCOT و کتولیز در موش های SCOT-KO نشان داده شد که به دلیل هیپوگلیسمی هیپکتونمی در طی 48h بعد از تولد (Coot et al.، 2011) نشان داده شد. از بین رفتن بافت SCOT در نورون ها یا میوسیت های اسکلتی باعث ایجاد اختلالات متابولیکی در هنگام گرسنگی می شود، اما کشنده نیست (Cotter et al.، 2013b). در انسان، کمبود SCOT در اوایل زندگی با کتوآکیدوز شدید ایجاد می کند، باعث بی نظمی، استفراغ و کما می شود (Berry و همکاران، 2001؛ Fukao و همکاران، 2000؛ Kassovska-Bratinova و همکاران، 1996؛ Niezen-Koning و همکاران. ، 1997؛ Saudubray و همکاران، 1987؛ Snyderman و همکاران، 1998؛ Tildon و Cornblath، 1972). نسبت سطح کمی در سطح سلولی در مورد ژن SCOT و تنظیم کننده های بیان پروتئین شناخته شده است. Expression of mRNA Oxct1 و پروتئین و فعالیت SCOT در حالت های کوتیوتیکی، احتمالا از طریق مکانیسم های وابسته به PPAR (Fenselau و Wallis، 1974، Fenselau و Wallis، 1976، Grinblat و همکاران، 1986، Okuda و همکاران، 1991، Turko et al .، 2001؛ Wentz و همکاران، 2010). در کتوآکیدوز دیابتی، عدم همبستگی بین کتوژنز کبدی و اکسیداسیون بیش از حد کبدی با اختلال فعالیت SCOT تشدید می شود. غلظت انسولین مستقل گلوکز (GLUT1 / SLC2A1) در كاردیومیوسیت ها همچنین بیان ژن Oxct1 را مهار می كند و اكسیداسیون تركیبی كاتون ها را در حالت غیر كتوتیكی (Yan et al.، 2009) پایین می آورد. در کبد، فراوانی mRNA Oxct1 توسط microRNA-122 و متیلاسیون هیستون H3K27me3 سرکوب می شود که در طی گذار از جنین به دوره نوزادی دیده می شود (Thorrez et al.، 2011). با این حال، سرکوب بیان Oxct1 کبدی در دوره پس از تولد در درجه اول به تخلیه پیش سازهای خونگرم بیان کننده Oxct1 از کبد، به جای از دست دادن بیان Expression Oxct1 که قبلا در hepatocytes differentiated پایدار وجود دارد. در واقع، بیان پروتئین Oxct1 mRNA و SCOT در hepatocytes های متمایز بسیار کم است (Orii et al.، 2008).
SCOT توسط PTM ها نیز تنظیم می شود. آنزیم در مغز موش های SIRT3 KO بیش از حد استیل شده است ، که همچنین تولید استیل-CoA وابسته به AcAc را نشان می دهد (Dittenhafer-Reed و همکاران ، 2015). نیتراتاسیون غیر آنزیمی باقیمانده تیروزین SCOT نیز فعالیت آن را کاهش می دهد ، که در قلب مدل های مختلف موش دیابتی گزارش شده است (مارکوندز و همکاران ، 2001 ، تورکو و دیگران ، 2001 ؛ وانگ و همکاران ، 2010a). در مقابل ، نیتراتاسیون باقیمانده تریپتوفان فعالیت SCOT را افزایش می دهد (Br g re et al.، 2010؛ Rebrin et al.، 2007). مکانیزم های مولکولی نیتراتاسیون یا نیتراتاسیون خاص باقیمانده که برای تعدیل فعالیت SCOT طراحی شده اند ممکن است وجود داشته باشد و نیاز به توضیح داشته باشد.
اختلافات در کتوز زخم ناخوشایند
در پستانداران اندام کتوژنیک اولیه کبد است و فقط سلولهای کبدی و سلولهای اپیتلیال روده ایزوفرم میتوکندری HMGCS2 را به طور فراوان بیان می کنند (Cotter et al.، 2013a؛ Cotter et al.، 2014؛ McGarry and Foster، 1980؛ Robinson and Williamson، 1980) . تخمیر باکتریایی بی هوازی از پلی ساکاریدهای پیچیده ، بوتیرات تولید می کند که برای اکسیداسیون یا کتوژنز توسط پستانداران توسط کولونوسیت ها جذب می شود (چربوی و همکاران ، 1995) ، که ممکن است در تمایز کولونوسیت ها نقش داشته باشد (وانگ و همکاران ، 2016). به استثنای سلولهای اپیتلیال روده و سلولهای کبدی ، HMGCS2 تقریباً در سایر سلولهای پستانداران وجود ندارد ، اما احتمال کتوژنز خارج کبدی در سلولهای تومور ، آستروسیتهای سیستم عصبی مرکزی ، کلیه ، لوزالمعده افزایش یافته است؟ سلولها ، اپیتلیوم رنگدانه شبکیه (RPE) و حتی در عضله اسکلتی (Adijanto et al.، 2014؛ Avogaro et al.، 1992؛ El Azzouny et al.، 2016؛ Grabacka et al.، 2016؛ Kang et al.، 2015 ؛ Le Foll و همکاران ، 2014 ؛ نوناکا و همکاران ، 2016 ؛ Takagi و همکاران ، 2016a ؛ Thevenet و همکاران ، 2016 ؛ Zhang و همکاران ، 2011). HMGCS2 خارج رحمی در بافتهایی مشاهده می شود که فاقد ظرفیت کتوژنیک خالص هستند (کوک و همکاران ، 2016 ؛ ونتز و همکاران ، 2010) و HMGCS2 فعالیتهای «نور مهتابی» مستقل از کتوژنز را نشان می دهد ، از جمله در هسته سلول (Chen و همکاران). ، 2016 ؛ Kostiuk و همکاران ، 2010 ؛ Meertens و همکاران ، 1998).
هر بافت خارج کبدی که اجزای کتون را اکسید می کند ، همچنین توانایی جمع شدن اجسام کتون از طریق مکانیزم های مستقل HMGCS2 را دارد (شکل 2A). با این حال ، هیچ بافت خارج کبدی وجود ندارد که در آن غلظت بدن کتون حالت پایدار بیش از آن باشد که در گردش خون وجود دارد (Cotter et al.، 2011؛ Cotter et al.، 2013b؛ Harrison and Long، 1940) ، با تأکید بر اینکه اجسام کتون به پایین منتقل می شوند شیب غلظت از طریق مکانیسم های وابسته به MCT1 / 2. یک مکانیسم کتوژنز خارج کبدی آشکارا ممکن است منعکس کننده اختلال نسبی اکسیداسیون کتون باشد. توضیحات بالقوه اضافی در حوزه تشکیل بدن کتون قرار دارد. اول ، کتوژنز نو ممکن است از طریق فعالیت آنزیمی برگشت پذیر تیولاز و SCOT رخ دهد (ویدمان و کربس ، 1969). وقتی غلظت استیل-CoA نسبتاً زیاد باشد ، واکنشهایی که معمولاً مسئول اکسیداسیون AcAc هستند در جهت معکوس عمل می کنند (GOLDMAN ، 1954). مکانیسم دوم زمانی اتفاق می افتد که واسطه های ناشی از اکسیداسیون؟ به دلیل تنگنای چرخه TCA جمع شوند ، AcAc-CoA از طریق یک واکنش کاتالیز شده توسط دهیدروژناز 3-هیدروکسی آسیل-CoA میتوکندری و بیشتر توسط 3-هیدروکسی بوتیریل به l-؟ OHB-CoA تبدیل می شود CoA deacylase به l-؟ OHB ، که با طیف سنجی جرمی یا طیف سنجی رزونانس از انانتیومر فیزیولوژیکی d-؟ OHB قابل تشخیص نیست (Reed and Ozand، 1980). l-؟ OHB را می توان از نظر كروماتوگرافی یا آنزیمی از d-؟ OHB تشخیص داد و در بافتهای خارج كبدی وجود دارد ، اما در كبد یا خون وجود ندارد (Hsu et al.، 2011). کتوژنز کبدی فقط d-؟ OHB ، تنها آنانتیومر که یک بستر BDH است ، تولید می کند (Ito et al.، 1984؛ Lincoln et al.، 1987؛ Reed and Ozand، 1980؛ Scofield et al.، 1982؛ Scofield et al.، 1982) سومین سازوکار مستقل از HMGCS2 از طریق کاتابولیسم اسیدهای آمینه ، به ویژه مکانیسم لوسین و لیزین ، d-؟ OHB تولید می کند. مکانیزم چهارم فقط به این دلیل آشکار است که به دلیل مصنوعی برچسب زنی است و بنابراین شبه کوکتوژنز نامیده می شود. این پدیده مربوط به برگشت پذیری واکنشهای SCOT و تیولاز است و می تواند به دلیل رقت ایزوتوپی ردیاب بدن کتون در بافت خارج کبدی ، باعث تخمین زیاد گردش بدن کتون شود (Des Rosiers و همکاران ، 1990 ؛ فینک و دیگران ، 1988) . با این وجود ، شبه کوکتوژنز ممکن است در اکثر زمینه ها قابل اغماض باشد (بیلی و همکاران ، 1990 ؛ کلر و همکاران ، 1978). یک شماتیک (شکل 2A) نشانگر یک روش مفید برای استفاده در حالی است که غلظت حالت پایدار بافتهای کتون را در نظر بگیرید.
recently کلیه اخیراً به عنوان یک اندام بالقوه کتوژنیک مورد توجه قرار گرفته است. در اکثریت قریب به اتفاق ایالت ها ، کلیه یک مصرف کننده خالص از اجسام کتون مشتق شده از کبد است ، که بدن کتون را از جریان خون دفع یا جذب می کند و کلیه به طور کلی مولد یا متمرکز کننده بدن کتون خالص نیست (رابینسون و ویلیامسون ، 1980). نویسندگان یک مطالعه کلاسیک نتیجه گرفتند که حداقل کتوژنیز کلیوی که در یک سیستم آزمایشی مصنوعی کمی شده است ، از نظر فیزیولوژیکی مربوط نیست (ویدمان و کربس ، 1969). اخیراً ، کتوژنز کلیه در مدلهای موش مبتلا به کمبود دیابت و اتوفاژی استنباط شده است ، اما به احتمال زیاد تغییرات چند عضوی در هموستاز متابولیکی ، متابولیسم کتون یکپارچه را از طریق ورودی به اندامهای مختلف تغییر می دهد (Takagi و همکاران ، 2016a ؛ Takagi و همکاران ، 2016b ؛ ژانگ و همکاران ، 2011). یکی از انتشارات اخیر ، کتوژنز کلیه را به عنوان مکانیسم محافظتی در برابر آسیب ایسکمی جریان خون مجدد در کلیه پیشنهاد کرده است (تران و همکاران ، 2016). غلظت مطلق حالت پایدار OHHB از عصاره های بافت کلیوی موش در 4 12 میلی متر reported گزارش شد. برای آزمایش اینکه آیا این قابل قبول بود ، ما غلظت OHB را در عصاره های کلیه از موش های تغذیه شده و 24 ساعته روزانه تعیین کردیم. غلظت های OHB سرم با fast 100 ساعت ناشتا از ~ 2 M به 24 میلی مولار افزایش یافت (شکل 2B) ، در حالی که غلظت های OHB حالت پایدار کلیه 100 M در حالت تغذیه تقریبی و فقط 1 میلی متر در حالت روزه 24 ساعته است (شکل). 2C E) ، مشاهدات سازگار با غلظتهایی که بیش از 45 سال پیش تعیین شده است (همز و بروسنان ، 1970). این احتمال وجود دارد که در حالت های کتوتیک ، اجسام کتون مشتق از کبد بتوانند از محافظت مجدد محافظت کنند ، اما شواهد مربوط به کتوژنز کلیه نیاز به اثبات بیشتر دارد. شواهد قانع کننده ای که از کتوژنز خارج کبدی واقعی پشتیبانی می کند در RPE ارائه شده است (Adijanto و همکاران ، 2014). این تحول متابولیکی جذاب پیشنهاد شده است که به طور بالقوه اجازه می دهد کتونهای مشتق از RPE به سلولهای گیرنده نوری یا مولر گلیا منتقل شوند ، که می تواند به بازسازی بخش خارجی گیرنده نوری کمک کند.
OHB به عنوان یک واسطه سیگنالینگ
اگرچه آنها از نظر انرژی غنی هستند ، اجسام کتونی نقش هشدار دهنده "غیر متعارف" را در هموستاز سلولی دارند (شکل 3) (نیومن و وردین ، 2014 ؛ روجاس-مورالس و همکاران ، 2016). به عنوان مثال ،؟ OHB کلاس های HDAC را مهار می کند ، که استیلاسیون هیستون را افزایش می دهد و در نتیجه باعث بیان ژن هایی می شود که استرس اکسیداتیو را کاهش می دهند (Shimazu et al.، 2013). ؟ OHB خود یک اصلاح کننده کووالانسی هیستون در باقیمانده های لیزین در کبد موش های دیابتی ناشتا یا استرپتوزوتوسین است (Xie و همکاران ، 2016) (همچنین به زیر مراجعه کنید ، ادغام متابولیسم بدن کتون ، اصلاح پس از ترجمه و فیزیولوژی سلول و اجسام کتون ، استرس اکسیداتیو و محافظت در برابر نورون).
�
؟ OHB همچنین از طریق گیرنده های همراه پروتئین G تأثیرگذار است. از طریق مکانیسم های نامشخص مولکولی ، فعالیت سیستم عصبی سمپاتیک را سرکوب می کند و با مهار سیگنالینگ اسیدهای چرب زنجیره کوتاه از طریق گیرنده 41 پروتئین G (GPR41) ، کل انرژی و ضربان قلب را کاهش می دهد (کیمورا و همکاران ، 2011). یکی از مهمترین اثرات سیگنالینگ؟ OHB از طریق GPR109A (همچنین به عنوان HCAR2 شناخته می شود) ، عضوی از خانواده فرعی هیدروکربوکسیلیک اسید GPCR که در بافتهای چربی (سفید و قهوه ای) بیان می شود ، حاصل می شود (تونارو و دیگران ، 2003) و در سلولهای ایمنی (احمد و همکاران ، 2009). ؟ OHB تنها لیگاند درون زا گیرنده GPR109A شناخته شده است (EC50 ~ 770 M) که توسط d-؟ OHB ، l-؟ OHB و بوتیرات فعال می شود ، اما AcAc نیست (Taggart و همکاران ، 2005). آستانه غلظت بالا برای فعال سازی GPR109A از طریق پیروی از رژیم کتوژنیک ، گرسنگی یا در طی کتواسیدوز به دست می آید ، که منجر به مهار لیپولیز بافت چربی می شود. اثر ضد لیپولیتیک GPR109A از طریق مهار آدنیلیل سیکلاز و کاهش اردوگاه ، مهار تری گلیسیرید لیپاز حساس به هورمون پیش می رود (احمد و همکاران ، 2009 ؛ تونارو و دیگران ، 2003). این یک حلقه بازخورد منفی ایجاد می کند که در آن کتوز با کاهش ترشح اسیدهای چرب غیراستری شده از سلولهای چربی ، ترمز تعدیلی را روی کتوژنز ایجاد می کند (احمد و همکاران ، 2009 ؛ تاگگارت و همکاران ، 2005) ، تاثیری که می تواند با آن تعادل داشته باشد محرک دلسوزانه که لیپولیز را تحریک می کند. نیاسین (ویتامین B3 ، اسید نیکوتینیک) یک لیگاند قوی (EC50 ~ 0.1 M) برای GRP109A است ، که به طور م forثر برای دهه ها برای اختلالات چربی استفاده می شود (بنیو و همکاران ، 2005 ؛ بنیو و همکاران ، 2006 ؛ Fabbrini و همکاران ، 2010a ؛ لوکاسووا و دیگران ، 2011 ؛ تونارو و دیگران ، 2003). در حالی که نیاسین انتقال کلسترول معکوس را در ماکروفاژها افزایش می دهد و ضایعات آترواسکلروتیک را کاهش می دهد (Lukasova et al.، 2011) ، اثرات OHH بر ضایعات آترواسکلروتیک ناشناخته باقی مانده است. اگرچه گیرنده GPR109A نقش محافظتی دارد و ارتباطات جذاب بین استفاده از رژیم کتوژنیک در سکته مغزی و بیماری های نورودژنراتیو وجود دارد (Fu et al.، 2015؛ Rahman et al.، 2014) ، نقش محافظتی از؟ OHB از طریق GPR109A در داخل بدن اثبات نشده است .
سرانجام ، OHB ممکن است اشتها و سیری را تحت تأثیر قرار دهد. متاآنالیز مطالعاتی که اثرات رژیم های کتوژنیک و بسیار کم انرژی را اندازه گیری می کند به این نتیجه رسیده است که شرکت کنندگان در این رژیم ها نسبت به رژیم های شاهد سیری بیشتری نشان می دهند (گیبسون و همکاران ، 2015). با این حال ، یک توضیح قابل قبول برای این اثر عناصر اضافی متابولیکی یا هورمونی است که ممکن است اشتها را تعدیل کند. به عنوان مثال ، موشهایی که تحت رژیم کتوژنیک جوندگان قرار داشتند ، با وجود دریافت کالری مشابه ، افزایش انرژی را در مقایسه با موشهای کنترل شده چو افزایش دادند و لپتین در گردش خون یا ژنهای پپتیدهای تنظیم کننده رفتار تغذیه ای تغییر نکردند (کندی و همکاران ، 2007). در میان مکانیسم های پیشنهادی که نشان می دهد سرکوب اشتها توسط؟ OHB شامل سیگنالینگ و اکسیداسیون است (Laeger et al.، 2010). حذف اختصاصی هپاتوسیت از ژن ریتم شبانه روزی (Per2) و مطالعات ایمن سازی کروماتین نشان داد که PER2 به طور مستقیم ژن Cpt1a را فعال می کند و به طور غیر مستقیم Hmgcs2 را تنظیم می کند و منجر به اختلال در کتوز در موش های حذفی Per2 می شود (Chavan و همکاران ، 2016). این موش ها پیش بینی غذایی مختل شده ای را نشان دادند که تا حدی با استفاده از سیستم سیستمیک OHH ترمیم شد. برای تأیید سیستم عصبی مرکزی به عنوان هدف مستقیم OHB ، و اینکه آیا اکسیداسیون کتون برای اثرات مشاهده شده مورد نیاز است یا اینکه مکانیسم سیگنالینگ دیگری درگیر است ، به مطالعات آینده نیاز است. محققان دیگر احتمال کتوژنز محلی مشتق شده از آستروسیت را در هیپوتالاموس ونترومدیال به عنوان تنظیم کننده دریافت غذا استناد کرده اند ، اما این مشاهدات اولیه نیز از ارزیابی های ژنتیکی و شار بهره مند می شوند (Le Foll و همکاران ، 2014). رابطه بین کتوز و محرومیت از عناصر غذایی همچنان مورد توجه است زیرا گرسنگی و سیری عناصر مهم در تلاشهای ناموفق کاهش وزن هستند.
ادغام متابولیسم بدن کتون، اصلاح Post-translational و فیزیولوژی سلولی
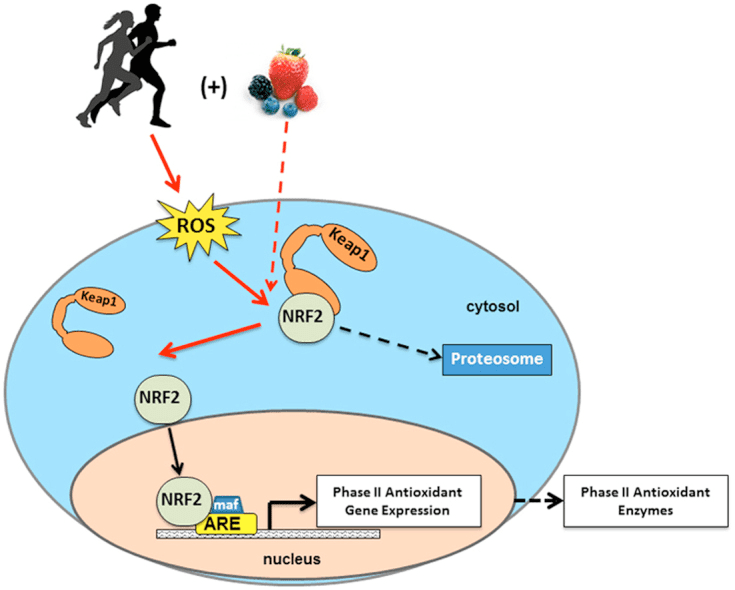
بدن کتون ها به استخرهای کم عمق استیل کوا کمک می کند که یک میان وعده کلیدی است که نقش کلیدی در متابولیسم سلولی را ایفا می کند (Pietrocola et al.، 2015). یکی از نقش استیل کولا، آن است که به عنوان یک سوبسترای برای استیل کردن، یک اصلاح کوانتومی هیستون با کاتالیزوری آنزیمی کاتالیز شود (Choudhary et al.، 2014؛ Dutta et al.، 2016؛ Fan et al.، 2015؛ Menzies et al.، 2016 ) تعداد زیادی از پروتئین های میتوکندری که به صورت پویا آتیلیتی شده اند و بسیاری از آنها ممکن است از طریق مکانیسم های غیر آنزیمی رخ دهد نیز از مطالعات پروتئومیک محاسباتی (Dittenhafer-Reed و همکاران، 2015، Hebert و همکاران، 2013، Rardin و همکاران، 2013 ؛ Shimazu و همکاران، 2010). Lysine deacetylases از کافاکر روی (به عنوان مثال، HDAC های نوکلئوسیتوزول) و یا NAD + به عنوان یک سوبسترا استفاده می کنند (sirtuins، SIRTs) (Choudhary و همکاران، 2014؛ Menzies و همکاران، 2016). استیل پروتئوم به عنوان سنسور و اثر بخشی از کل استیل کولا سلولی عمل می کند، به نحوی که دستکاری های ژنتیکی و فیزیولوژیکی هر کدام منجر به تغییرات جهانی آنزیمی استیلینگ می شود (Weinert et al.، 2014). از آنجایی که متابولیتهای داخل سلولی به عنوان مدولاتورهای استیلایزر باقی مانده لیزین عمل می کنند، مهم است که نقش کتون ها، که فراوانی بسیار پویا هستند، مورد توجه قرار گیرد.
؟ OHB یک اصلاح کننده اپی ژنتیک است که حداقل از طریق دو مکانیزم انجام می شود. افزایش؟ سطح OHB ناشی از روزه داری ، محدودیت کالری ، تجویز مستقیم یا ورزش طولانی مدت باعث مهار HDAC یا فعال سازی استیل ترانسفراز هیستون می شود (ماروسی و همکاران ، 2016 ؛ سلیمان و همکاران ، 2016) یا به استرس اکسیداتیو (Shimazu و همکاران ، 2013) . مهار OHB از HDAC3 می تواند فیزیولوژی متابولیک نوزاد را تنظیم کند (Rando و همکاران ، 2016). به طور مستقل ،؟ OHB خود مستقیماً بقایای هیستون لیزین را اصلاح می کند (زی و دیگران ، 2016). روزه داری طولانی مدت یا کتواسیدوز دیابتی ناشی از استپتوزوتوسین باعث افزایش بتا-هیدروکسی بوتیریلاسیون هیستون می شود. اگرچه تعداد سایت های بتا-هیدروکسی بوتیریلاسیون و استیلاسیون لیزین قابل مقایسه بود ، اما از لحاظ استوکیومتری هیستون؟ -هیدروکسی بوتیریلاسیون بیشتر از استیلاسیون مشاهده شد. ژنهای متمایز تحت تأثیر آلفا-هیدروکسی بوتیریلاسیون هیستون لیزین ، در مقابل استیلاسیون یا متیلاسیون تحت تأثیر قرار گرفتند ، که عملکردهای سلولی متمایز را نشان می دهد. آیا هیدروکسی بوتیریلاسیون خود به خود است یا آنزیمی مشخص نیست ، اما دامنه مکانیسم ها را از طریق اجسام کتون به طور پویا رونویسی را تحت تأثیر قرار می دهد.
حوادث برنامه ریزی مجدد سلول ضروری در طول محدودیت کالری و محرومیت از مواد مغذی ممکن است در ازدیسیلاسیون میتوکندری و desuccinylation وابسته به SIRT3 و SIRT5 ، به ترتیب ، تنظیم پروتئین های کتوژنیک و کتولیتیک در سطح پس از ترجمه در بافت های کبدی و خارج کبدی (Dittenhafer-Reed و همکاران ، 2015 ؛ هبرت و همکاران ، 2013 ؛ راردین و همکاران ، 2013 ؛ شیمازو و همکاران ، 2010). حتی اگر مقایسه استوکیومتری مکانهای اشغال شده لزوماً مستقیماً به تغییر در شار متابولیکی ارتباط نداشته باشد ، استیلاسیون میتوکندری پویا است و ممکن است به جای استیل ترانسفرازهای آنزیمی توسط غلظت استیل-CoA یا pH میتوکندری هدایت شود (واگنر و پین ، 2013). اینکه SIRT3 و SIRT5 فعالیتهای آنزیمهای متابولیسم بدن کتون را تعدیل می کنند ، این س questionال را نشان می دهد که نقش متقابل کتونها در مجسمه سازی استیل پروتئین ، سوکسینیل پروتئین و سایر اهداف سلولی پویا چیست. در واقع ، همانطور که تغییرات کتوژنز منعکس کننده غلظت NAD + است ، تولید کتون و فراوانی آن می تواند فعالیت سیرتین را تنظیم کند ، در نتیجه بر استخرهای کل استیل-CoA / succinyl-CoA ، آسیل پروتئین و در نتیجه فیزیولوژی میتوکندری و سلول تأثیر می گذارد. ؟ -هیدروکسی بوتیریلاسیون باقیمانده آنزیم لیزین می تواند لایه دیگری به برنامه ریزی مجدد سلول اضافه کند. در بافتهای خارج کبدی ، اکسیداسیون بدن کتون ممکن است تغییرات مشابه در هموستاز سلول را تحریک کند. در حالی که محفظه استخرهای استیل-CoA بسیار تنظیم شده و طیف گسترده ای از تغییرات سلولی را هماهنگ می کند ، توانایی اجسام کتون در شکل دادن مستقیم غلظت استیل-CoA میتوکندری و سیتوپلاسمی به توضیح نیاز دارد (چن و همکاران ، 2012 ؛ Corbet و همکاران ، 2016 ؛ پوگوکوینا و دیگران ، 2014 ؛ شوور و همکاران ، 2009 ؛ ولن و تامپسون ، 2012). از آنجا که غلظت استیل-CoA به شدت تنظیم می شود ، و استیل-CoA غشا و فرآیندهای غشایی نفوذ ناپذیر است ، در نظر گرفتن مکانیسم های محرک هماهنگی هموستاز استیل-CoA از جمله میزان تولید و اکسیداسیون ترمینال در چرخه TCA ، تبدیل به اجسام کتون ، میتوکندری بسیار مهم است. پساب از طریق کارنیتین استیل ترانسفراز (CrAT) یا صادرات استیل-CoA به سیتوزول پس از تبدیل به سیترات و انتشار توسط ATP سیترات لیاز (ACLY). نقشهای اصلی این مکانیزمهای اخیر در استیل پروتئین سلول و هموستاز نیاز به درک منطقی نقش کتوژنز و اکسیداسیون کتون دارد (Das et al.، 2015؛ McDonnell et al.، 2016؛ Moussaieff et al.، 2015؛ Overmyer et al.) 2015 ؛ Seiler و همکاران ، 2014 ؛ Seiler و همکاران ، 2015 ؛ Wellen و همکاران ، 2009 ؛ Wellen و Thompson ، 2012). برای تعیین اهداف و نتایج ، به فن آوری های همگرا در متابولومیک و آسیلپروتئومیک در تنظیم مدل های دستکاری شده ژنتیکی نیاز خواهد بود.
پاسخ های ضد و ضد التهابی به بدن کتون
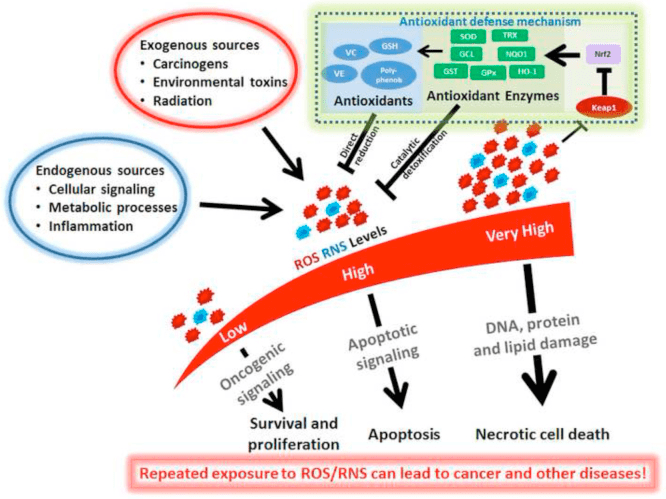
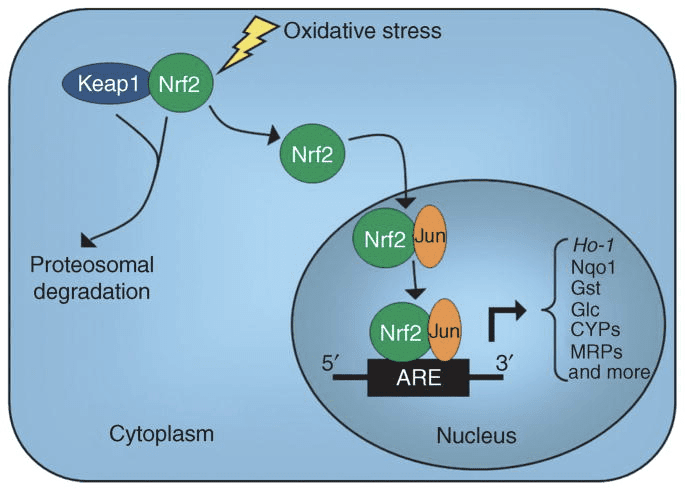
کتوز و اجسام کتون التهاب و عملکرد سلولهای ایمنی را تعدیل می کنند ، اما مکانیسم های متنوع و حتی متناقضی ارائه شده است. کمبود طولانی مدت مواد مغذی باعث کاهش التهاب می شود (یوم و همکاران ، 2015) ، اما کتوز مزمن دیابت نوع 1 یک حالت پیش التهابی است (Jain et al.، 2002؛ Kanikarla-Marie and Jain، 2015؛ Kurepa et al.، 2012 ) نقش های سیگنالینگ مبتنی بر مکانیسم برای؟ OHB در التهاب ظاهر می شود زیرا بسیاری از سلول های سیستم ایمنی بدن ، از جمله ماکروفاژها یا مونوسیت ها ، به طور فراوان GPR109A را بیان می کنند. در حالی که؟ OHB عمدتا یک واکنش ضد التهابی اعمال می کند (Fu و همکاران ، 2014 ؛ Gambhir و همکاران ، 2012 ؛ رحمان و همکاران ، 2014 ؛ Youm و همکاران ، 2015) ، غلظت های بالای بدن کتون ، به ویژه AcAc ، ممکن است پاسخ پیش التهابی را ایجاد کنید (Jain et al.، 2002؛ Kanikarla-Marie and Jain، 2015؛ Kurepa et al.، 2012).
نقش ضد التهابی لیگاندهای GPR109A در تصلب شرایین ، چاقی ، بیماری التهابی روده ، بیماری عصبی و سرطان بررسی شده است (Graff و همکاران ، 2016). بیان GPR109A در سلولهای RPE مدلهای دیابتی ، بیماران دیابتی انسان (Gambhir و همکاران ، 2012) و در میکروگلیا در طی تخریب سلولهای عصبی افزایش می یابد (Fu و همکاران ، 2014). اثرات ضد التهابی؟ OHB با بیان بیش از حد GPR109A در سلولهای RPE افزایش یافته و با مهار دارویی یا حذفی ژنتیکی GPR109A لغو می شود (گامبیر و همکاران ، 2012). ؟ OHB و اسید نیکوتینیک برون زا (Taggart و همکاران ، 2005) ، هر دو اثرات ضد التهابی در TNF ایجاد می کنند؟ یا التهاب ناشی از LPS با کاهش سطح پروتئین های پیش التهاب (iNOS ، COX-2) یا سیتوکین های ترشحی (TNF ؟، IL-1 ؟، IL-6، CCL2 / MCP-1) ، تا حدی از طریق مهار NF - انتقال B (Fu و همکاران ، 2014 ؛ گامبیر و همکاران ، 2012). ؟ OHB استرس ER و التهاب NLRP3 را کاهش می دهد و پاسخ استرس آنتی اکسیدانی را فعال می کند (Bae et al.، 2016؛ Youm et al.، 2015). با این حال ، در التهاب نورودژنراتیو ، محافظت با واسطه وابسته به GPR109A شامل واسطه های التهابی مانند سیگنالینگ مسیر MAPK (به عنوان مثال ، ERK ، JNK ، p38) نیست (Fu et al.، 2014) ، اما ممکن است به PGD1 وابسته به COX-2 نیاز داشته باشد تولید (رحمان و همکاران ، 2014). جالب است که ماکروفاژ GPR109A برای اعمال اثر محافظت عصبی در مدل سکته مغزی ایسکمیک مورد نیاز است (رحمان و همکاران ، 2014) ، اما توانایی؟ OHB برای مهار التهاب NLRP3 در ماکروفاژهای مشتق از مغز استخوان مستقل از GPR109A است (Youm و همکاران . ، 2015). اگرچه بیشتر مطالعات ، OHB را به اثرات ضد التهابی مرتبط می دانند ، اما OHB ممکن است پیش التهاب باشد و مارکرهای پراکسیداسیون لیپید را در سلولهای کبدی گوساله افزایش دهد (Shi et al.، 2014). اثرات ضد- در مقابل التهابی؟ OHB ممکن است به نوع سلول ، غلظت؟ OHB ، مدت قرار گرفتن در معرض ، و وجود یا عدم تعدیل کننده های مشترک بستگی داشته باشد.
بر خلاف؟ OHB ، AcAc ممکن است سیگنالینگ پیش التهابی را فعال کند. افزایش AcAc ، به ویژه با غلظت گلوکز بالا ، آسیب سلول های اندوتلیال را از طریق مکانیسم NADPH اکسیداز / استرس اکسیداتیو تشدید می کند (Kanikarla-Marie و Jain ، 2015). غلظت بالای AcAc در بند ناف مادران دیابتی با میزان اکسیداسیون پروتئین بالاتر و غلظت MCP-1 ارتباط داشت (Kurepa و همکاران ، 2012). AcAc بالا در بیماران دیابتی با TNF ارتباط داشت؟ بیان (Jain و همکاران ، 2002) ، و AcAc ، اما نه؟ OHB ، بیان TNF؟ ، MCP-1 ، تجمع ROS ، و سطح اردوگاه در سلولهای مونوسیت انسانی U937 کاهش می یابد (Jain و همکاران ، 2002 ؛ Kurepa و همکاران . ، 2012).
پدیده های سیگنالینگ وابسته به بدن کتون غالباً فقط با غلظت بالای بدن کتون (> 5 میلی متر) و در مورد بسیاری از مطالعات که کتون ها را به اثرات پیش التهابی یا ضد التهابی مرتبط می کند ، از طریق مکانیسم های نامشخص تحریک می شوند. علاوه بر این ، با توجه به اثرات متناقض؟ OHB در مقابل AcAc بر التهاب ، و توانایی نسبت AcAc /؟ OHB در تأثیر پتانسیل اکسایش میتوکندری ، بهترین آزمایش ارزیابی نقش اجسام کتون بر فنوتیپ های سلولی مقایسه اثرات AcAc و؟ OHB در نسبت های مختلف و در غلظت های مختلف تجمعی [به عنوان مثال ، (Saito et al.، 2016)]. سرانجام ، AcAc را می توان از طریق تجاری فقط به عنوان نمک لیتیوم یا به عنوان یک استر اتیل که قبل از استفاده به هیدرولیز پایه نیاز دارد ، خریداری کرد. کاتیون لیتیوم به طور مستقل باعث ایجاد آبشارهای انتقال سیگنال می شود (مانجی و همکاران ، 1995) و آنیون AcAc ناپایدار است. سرانجام ، مطالعات با استفاده از d / l-؟ OHB نژادی می تواند اشتباه گرفته شود ، زیرا فقط استریوایزومر d-؟ OHB می تواند به AcAc اکسید شود ، اما d-؟ OHB و l-؟ OHB می تواند هر یک از طریق GPR109A سیگنال داده ، التهاب NLRP3 را مهار کند ، و به عنوان بسترهای لیپوژنیک خدمت می کنند.
بدن کتون، استرس اکسیداتیو، و محافظت از عصبی
استرس اکسیداتیو معمولاً به عنوان حالتی تعریف می شود که ROS به دلیل تولید بیش از حد و / یا از بین بردن اختلال ، بیش از حد ارائه شود. نقش های کاهش دهنده استرس آنتی اکسیدانی و اکسیداتیو اجسام کتون به طور گسترده ای هم در شرایط in vitro و هم در داخل بدن ، خصوصاً در زمینه محافظت از نور ، توصیف شده است. از آنجا که اکثر سلولهای عصبی فسفاتهای پر انرژی از اسیدهای چرب به طور مثر تولید نمی کنند ، اما در صورت کمبود کربوهیدرات ها ، بدن کتون را اکسید می کنند ، اثرات محافظت از نور در بدن کتون به ویژه مهم است (Cahill GF Jr، 2006؛ Edmond et al.، 1987؛ Yang) و دیگران ، 1987). در مدل های استرس اکسیداتیو ، القای BDH1 و سرکوب SCOT نشان می دهد که متابولیسم بدن کتون می تواند برای حفظ سیگنالینگ سلول های متنوع ، پتانسیل اکسایش اکسیداسیون یا نیازهای متابولیکی ، دوباره برنامه ریزی شود (Nagao et al.، 2016؛ Tieu et al.، 2003).
اجسام کتون باعث کاهش درجه آسیب سلولی ، آسیب ، مرگ و کاهش آپوپتوز در سلولهای عصبی و قلب می شوند (Haces et al.، 2008؛ Maalouf et al.، 2007؛ Nagao et al.، 2016؛ Tieu et al.، 2003). مکانیسم های فراخوانی شده متنوع هستند و همیشه به طور خطی با غلظت ارتباط ندارند. غلظت میلی مولار پایین (d یا l) -؟ OHB ROS ROS (آنیون هیدروکسیل) ، در حالی که AcAc گونه های مختلف ROS را از بین می برد ، اما فقط در غلظت هایی که بیش از حد فیزیولوژیکی باشد (IC50 20-67 میلی متر) (Haces و همکاران ، 2008) . برعکس ، یک نفوذ مفید بر پتانسیل اکسایش اکسیداسیون زنجیره انتقال الکترون مکانیزمی است که معمولاً با d-؟ OHB مرتبط است. در حالی که هر سه بدن کتون (d / l-؟ OHB و AcAc) باعث کاهش مرگ سلول های عصبی و تجمع ROS ناشی از مهار شیمیایی گلیکولیز می شوند ، فقط d-؟ OHB و AcAc مانع از کاهش ATP عصبی می شوند. برعکس ، در یک مدل کاهش قند خون در داخل بدن ، (d یا l) -؟ OHB ، اما AcAc از پراکسیداسیون لیپید هیپوکامپ جلوگیری نمی کند (Haces et al.، 2008؛ Maalouf et al.، 2007؛ Marosi et al.، 2016؛ Murphy، 2009 ؛ Tieu و همکاران ، 2003). مطالعات in vivo بر روی موش هایی که از رژیم کتوژنیک تغذیه می کردند (87٪ کیلو کالری چربی و 13٪ پروتئین) ، تغییرات عصبی آناتومیکی ظرفیت آنتی اکسیدانی را نشان دادند (زیگلر و همکاران ، 2003) ، که بیشترین تغییرات عمیق در هیپوکامپ مشاهده شد ، با افزایش گلوتاتیون پراکسیداز و کل ظرفیت آنتی اکسیدان
رژیم کتوژنیک ، استرهای کتونی (همچنین به استفاده درمانی از رژیم کتوژنیک و اجسام کتون برونزا مراجعه کنید) ، یا دولت OHB از محافظت عصبی در مدل های سکته مغزی ایسکمیک استفاده می کند (رحمان و همکاران ، 2014). بیماری پارکینسون (Tieu و همکاران ، 2003) ؛ سیستم عصبی مرکزی تشنج سمیت اکسیژن (D'Agostino و همکاران ، 2013) ؛ اسپاسم صرع (یوم و همکاران ، 2015) ؛ انسفالومیوپاتی میتوکندریایی ، اسیدوز لاکتیک و سندرم اپیزودهای مشابه سکته مغزی (MELAS) (فری و همکاران ، 2016) و بیماری آلزایمر (Cunnane و Crawford ، 2003 ؛ یین و همکاران ، 2016). برعکس ، یک گزارش اخیر شواهد هیستوپاتولوژیک از پیشرفت نورودژنراتیو توسط یک رژیم کتوژنیک را در یک مدل موش تراریخته از ترمیم DNA غیر طبیعی میتوکندری ، با وجود افزایش در بیوژنز میتوکندری و امضای آنتی اکسیدان نشان داده است (Lauritzen و همکاران ، 2016). گزارش های متناقض دیگر حاکی از آن است که قرار گرفتن در معرض غلظت بالای بدن کتون باعث ایجاد استرس اکسیداتیو می شود. دوزهای بالا؟ OHB یا AcAc باعث ترشح اکسید نیتریک ، پراکسیداسیون لیپید ، کاهش بیان SOD ، گلوتاتیون پراکسیداز و کاتالاز در سلولهای کبدی گوساله می شود ، در حالی که در سلولهای کبدی موش صحرایی القای مسیر MAPK به AcAc نسبت داده می شود اما نه؟ OHB (Abdelmegeed و همکاران ، 2004) ؛ Shi و همکاران ، 2014 ؛ Shi و همکاران ، 2016).
روی هم رفته ، اکثر گزارشات ، OHH را به کاهش استرس اکسیداتیو مرتبط می کنند ، زیرا تجویز آن از تولید ROS / سوپراکسید جلوگیری می کند ، از پراکسیداسیون لیپید و اکسیداسیون پروتئین جلوگیری می کند ، سطح پروتئین آنتی اکسیدان را افزایش می دهد ، و تنفس میتوکندری و تولید ATP را بهبود می بخشد (عبدالمجید و دیگران ، 2004 ؛ Haces et al.، 2008؛ Jain et al.، 1998؛ Jain et al.، 2002؛ Kanikarla-Marie and Jain، 2015؛ Maalouf et al.، 2007؛ Maalouf and Rho، 2008؛ Marosi et al.، 2016؛ Tieu و همکاران ، 2003 ؛ یین و همکاران ، 2016 ؛ زیگلر و همکاران ، 2003). در حالی که AcAc با القای استرس اکسیداتیو ارتباط مستقیم تری نسبت به؟ OHB داشته است ، این تأثیرات همیشه به راحتی از پاسخ های احتمالی پیش التهابی جدا نمی شوند (Jain et al.، 2002؛ Kanikarla-Marie and Jain، 2015؛ Kanikarla-Marie و جین ، 2016). علاوه بر این ، بسیار مهم است که در نظر بگیریم که سود آنتی اکسیدانی آشکار ناشی از جیره های کتوژنیک پلیوتروپیک ممکن است توسط بدن کتون ها منتقل نشود و محافظت از سلول های عصبی توسط اجسام کتون ممکن است به طور کامل ناشی از استرس اکسیداتیو نباشد. به عنوان مثال در طول محرومیت از گلوکز ، در یک مدل از محرومیت از گلوکز در سلول های عصبی قشر مغز ،؟ OHB شار اتوفاژی را تحریک می کند و از تجمع اتوفاگوزوم جلوگیری می کند ، که با کاهش مرگ عصبی همراه است (کامبروس-لونا و همکاران ، 2016). d-؟ OHB همچنین از طریق مهار HDAC باعث ایجاد پروتئین های آنتی اکسیدانی متعارف FOXO3a ، SOD ، MnSOD و کاتالاز می شود (Nagao et al.، 2016؛ Shimazu et al.، 2013).
بیماری کبد چرب غیر الکلی (NAFLD) و متابولیسم بدن کتون
NAFLD مرتبط با چاقی و استئو هپاتیت غیر الکلی (NASH) شایعترین علل بیماری کبدی در کشورهای غربی هستند (Rinella and Sanyal، 2016) و نارسایی کبدی ناشی از NASH یکی از دلایل عمده پیوند کبد است. در حالی که ذخیره بیش از حد تری اسیل گلیسرولها در سلولهای کبدی> 5٪ از وزن کبد (NAFL) به تنهایی باعث عملکرد تخریب کبد نمی شود ، پیشرفت در NAFLD در انسان با مقاومت به انسولین سیستمیک و افزایش خطر ابتلا به دیابت نوع 2 ارتباط دارد و ممکن است در پاتوژنز بیماری قلبی عروقی و بیماری مزمن کلیه (Fabbrini et al.، 2009؛ Targher et al.، 2010؛ Targher and Byrne، 2013). مکانیسم های بیماری زا NAFLD و NASH کاملاً شناخته نشده اند اما شامل ناهنجاری های متابولیسم کبدی ، اتوفاژی سلولهای کبدی و استرس شبکه آندوپلاسمی ، عملکرد سلولهای ایمنی کبدی ، التهاب بافت چربی و واسطه های التهابی سیستمیک هستند (Fabbrini et al.، 2009؛ Masuoka and Chalasani، 2013 ؛ Targher و همکاران ، 2010 ؛ یانگ و همکاران ، 2010). اغتشاشات کربوهیدرات ، لیپید و متابولیسم اسیدهای آمینه در انسان و در ارگانیسم های مدل ایجاد می شود و به چاقی ، دیابت و NAFLD کمک می کند [بررسی شده در (Farese et al.، 2012؛ Lin and Accili، 2011؛ Newgard، 2012؛ Samuel and شولمن ، 2012 ؛ خورشید و لازار ، 2013)]. در حالی که ناهنجاری های سلولهای کبدی در متابولیسم لیپیدهای سیتوپلاسمی معمولاً در NAFLD مشاهده می شود (Fabbrini et al.، 2010b) ، نقش متابولیسم میتوکندریا ، حاکم بر دفع اکسیداتیو چربی ها ، در پاتوژنز NAFLD کمتر مشخص است. ناهنجاری های متابولیسم میتوکندری در پاتوژنز NAFLD / NASH اتفاق می افتد و به آن کمک می کند (Hyotylainen et al.، 2016؛ Serviddio et al.، 2011؛ Serviddio et al.، 2008؛ Wei et al.، 2008). کلی وجود دارد (Felig و همکاران ، 1974 ؛ Iozzo و همکاران ، 2010 ؛ کولیاکی و همکاران ، 2015 ؛ Satapati و همکاران ، 2015 ؛ Satapati و همکاران ، 2012 ؛ Sunny و همکاران ، 2011) اما یکنواخت نیست ( کولیاکی و رودن ، 2013 ؛ پری و همکاران ، 2016 ؛ رکتور و همکاران ، 2010) اتفاق نظر داشتند که ، قبل از توسعه NASH با ایمان ، اکسیداسیون میتوکندری کبدی ، و به ویژه اکسیداسیون چربی ، در چاقی ، مقاومت انسولین سیستمیک افزایش می یابد ، و NAFLD. این احتمال وجود دارد که با پیشرفت NAFLD ، ناهمگنی ظرفیت اکسیداتیو ، حتی در میان میتوکندری های فردی ، ظاهر شود و در نهایت عملکرد اکسیداتیو مختل شود (کولیاکی و همکاران ، 2015 ؛ رکتور و دیگران ، 2010 ؛ Satapati و همکاران ، 2008 ؛ Satapati و همکاران . ، 2012).
کتوژنز اغلب به عنوان پروکسی برای اکسیداسیون چربی کبد استفاده می شود. با پیشرفت NAFLD در مدل های حیوانی و احتمالاً در انسان ، اختلالات کتوژنز ظاهر می شود. از طریق مکانیسم های کاملاً مشخص نشده ، هایپرینسولینمی کتوژنز را سرکوب می کند ، احتمالاً در مقایسه با گروه های کنترل نابارور در هیپوکتونمی نقش دارد (Bergman et al.، 2007؛ Bickerton et al.، 2008؛ Satapati et al.، 2012؛ Soeters et al.، 2009؛ Sunny et al. ، 2011 ؛ معاون و همکاران ، 2005). با این وجود ، توانایی غلظت بدن کتون در گردش برای پیش بینی NAFLD بحث برانگیز است (M nnist et al.، 2015؛ Sanyal et al.، 2001). روشهای طیف سنجی رزونانس مغناطیسی کمی قوی در مدلهای حیوانی ، میزان گردش کتون با مقاومت متوسط به انسولین را افزایش داد ، اما با مقاومت به انسولین شدید ، میزان کاهش مشهود بود (Satapati et al.، 2012؛ Sunny et al.، 2010). در انسانهای چاق با کبد چرب ، میزان کتوژنیک طبیعی است (Bickerton et al.، 2008؛ Sunny et al.، 2011) ، و از این رو ، میزان کتوژنز نسبت به افزایش بار اسیدهای چرب در سلولهای کبدی کاهش می یابد. در نتیجه ، استیل-CoA مشتق شده از اکسیداسیون ممکن است به اکسیداسیون ترمینال در چرخه TCA ، افزایش اکسیداسیون ترمینال ، گلوکونئوژنز هدایت شده توسط فسفوآنولپیروات از طریق آناپلروز / کاتاپلروز و استرس اکسیداتیو هدایت شود. استیل-CoA همچنین ممکن است تحت عنوان سیترات ، یک بستر پیش ساز برای لیپوژنز ، از میتوکندری صادر شود (شکل 4) (Satapati et al.، 2015؛ Satapati et al.، 2012؛ Solinas et al.، 2015). در حالی که کتوژنز با چاقی طولانی مدت کمتر به انسولین پاسخ می دهد یا ناشتا است (Satapati et al.، 2012) ، مکانیسم های اساسی و عواقب پایین دست این کاملاً شناخته نشده است. شواهد اخیر نشان می دهد که mTORC1 کتوژنز را به روشی که ممکن است در پایین دست سیگنالینگ انسولین باشد سرکوب می کند (Kucejova و همکاران ، 2016) ، که با مشاهداتی که mTORC1 از القای Hmgcs2 با واسطه PPAR جلوگیری می کند همخوانی دارد (Sengupta و همکاران ، 2010) ( همچنین به مقررات HMGCS2 و SCOT / OXCT1 مراجعه کنید).
�
مشاهدات اولیه از گروه ما پیامدهای نامطلوب کبدی ناشی از نارسایی کتوژنیک را نشان می دهد (Cotter et al.، 2014). برای آزمایش این فرضیه که اختلال کتوژنیز ، حتی در حالت پر از کربوهیدرات و در نتیجه "غیر کتوژنیک" ، به متابولیسم گلوکز غیر طبیعی کمک می کند و استئو کبدی را تحریک می کند ، ما با استفاده از اولیگونوکلئوتیدهای ضد عفونی (ASO) با استفاده از یک الگونوکلئوتید ضد عفونی کننده ، یک مدل موش را تولید کردیم. Hmgcs2 از دست دادن HMGCS2 در موشهای بزرگسال استاندارد تغذیه شده با چربی کم باعث افزایش قند خون خفیف و افزایش قابل توجه تولید صدها متابولیت کبدی می شود ، مجموعه ای که به شدت فعال سازی لیپوژنز را پیشنهاد می کند. تغذیه رژیم غذایی با چربی بالا از موش های دارای کتوژنیس کافی منجر به آسیب و التهاب گسترده سلولهای کبدی شد. این یافته ها فرضیه های اصلی را پشتیبانی می کند که (ک) کتوژنز یک مسیر سرریز غیرفعال نیست بلکه یک گره پویا در هموستاز فیزیولوژیکی کبدی و یکپارچه است و (ب) افزایش کتوژنیک محتاطانه برای کاهش NAFLD / NASH و متابولیسم گلوکز کبدی بی نظیر است و قابل بررسی است. .
کتوژنز مختل شده چگونه ممکن است به آسیب کبدی و تغییر هموستاز گلوکز کمک کند؟ اولین بررسی این است که آیا مقصر کمبود شار کتوژنیک است یا خود کتون ها. یک گزارش اخیر نشان می دهد که بدن کتون ممکن است آسیب کبدی ناشی از استرس اکسیداتیو را در پاسخ به اسیدهای چرب اشباع نشده n-3 کاهش دهد (Pawlak et al.، 2015). به یاد بیاورید که به دلیل عدم بیان SCOT در سلولهای کبدی ، اجسام کتون اکسید نمی شوند ، اما می توانند در لیپوژنز نقش داشته باشند ، و نقشهای مختلف سیگنالینگ را مستقل از اکسیداسیون آنها انجام می دهند (همچنین به سرنوشت های متابولیکی غیر اکسیداتیو بدن کتون و؟ OHB مراجعه کنید) یک واسطه سیگنالینگ). همچنین ممکن است اجسام کتونی مشتق شده از سلولهای کبدی به عنوان یک سیگنال و / یا متابولیت برای سلولهای همسایه در آکینوس کبدی ، از جمله سلولهای ستاره ای و ماکروفاژهای سلول کوپفر عمل کنند. در حالی که ادبیات محدود موجود نشان می دهد که ماکروفاژها قادر به اکسیداسیون اجسام کتون نیستند ، این تنها با استفاده از روش های کلاسیک و فقط در ماکروفاژهای صفاقی اندازه گیری شده است (Newsholme et al.، 1986؛ Newsholme et al.، 1987) ، نشان می دهد که یک ارزیابی با توجه به بیان فراوان SCOT در ماکروفاژهای مشتق شده از مغز استخوان مناسب است (یوم و همکاران ، 2015).
شار کتوژنیک هپاتوسیت نیز ممکن است از سلول محافظت کند. گرچه ممکن است مکانیسم های درمانی به خودی خود به کتوژنز وابسته نباشند ، رژیم های غذایی کتوژنیک با کربوهیدرات کم با بهبود NAFLD همراه بوده اند (براونینگ و همکاران ، 2011 ؛ فاستر و همکاران ، 2010 ؛ کانی و همکاران ، 2014 ؛ شوگر و کرافورد ، 2012) . مشاهدات ما نشان می دهد که کتوژنز سلولهای کبدی ممکن است شار چرخه TCA ، شار آناپلروتیک ، گلوکونئوژنز مشتق شده از فسفوآنولپیروات را بازخورد و تنظیم کند (Cotter et al.، 2014) و حتی گردش گلیکوژن. اختلال کتوژنیک استیل-CoA را به سمت افزایش شار TCA هدایت می کند ، که در کبد با افزایش آسیب ناشی از ROS مرتبط است (Satapati et al.، 2015؛ Satapati et al.، 2012). انحراف کربن را به سمت گونه های چربی سنتز شده جدید انجام می دهد که می تواند سمیت سلولی را ثابت کند. و از اکسیداسیون مجدد NADH به NAD + جلوگیری می کند (Cotter و همکاران ، 2014) (شکل 4). روی هم رفته ، آزمایشات بعدی برای حل سازوکارهایی لازم است که از طریق آنها نارسایی کتوژنیک نسبی ممکن است ناسازگار شود ، به افزایش قند خون کمک کند ، استئو هپاتیت را تحریک کند و اینکه آیا این مکانیسم ها در NAFLD / NASH انسان عمل می کنند. همانطور که شواهد اپیدمیولوژیک نشان دهنده اختلال در کتوژنیز در طی پیشرفت استئاتوپاتیت است (Embade et al.، 2016؛ Marinou et al.، 2011؛ M nnist et al.، 2015؛ Pramfalk et al.، 2015؛ Safaei et al.، 2016) درمانهایی که کتوژنز کبدی را افزایش می دهند می توانند مفید باشند (Degirolamo et al.، 2016؛ Honda et al.، 2016).
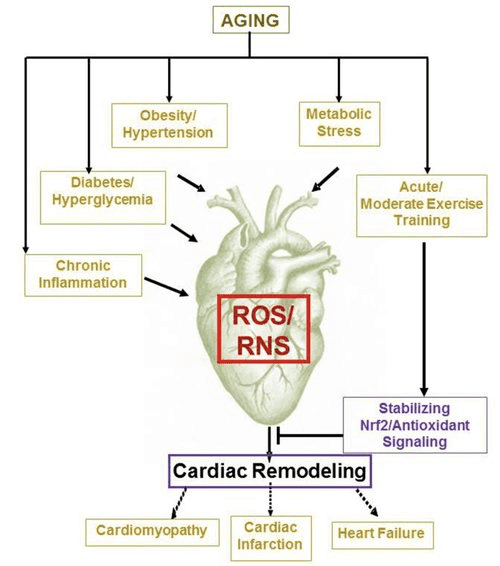
بدن کتون و نارسایی قلب (HF)
با سرعت متابولیسم بیش از 400 کیلوکالری در کیلوگرم در روز و گردش مالی 6-35 کیلوگرم ATP در روز ، قلب عضوی با بیشترین میزان مصرف انرژی و تقاضای اکسیداتیو است (اشرفیان و همکاران ، 2007 ؛ وانگ و همکاران ، 2010b) اکثریت قریب به اتفاق گردش انرژی میوکارد در میتوکندری ساکن است و 70٪ از این منبع از FAO سرچشمه می گیرد. قلب در شرایط عادی همه چیزخوار و انعطاف پذیر است ، اما بازسازی پاتولوژیک قلب (به عنوان مثال ، به دلیل فشار خون بالا یا سکته قلبی) و قلب دیابتی هر یک از نظر متابولیکی انعطاف پذیر نمی شوند (Balasse and Fery، 1989؛ BING، 1954؛ Fukao et al.، 2004 ؛ Lopaschuk و همکاران ، 2010 ؛ Taegtmeyer و همکاران ، 1980 ؛ Taegtmeyer و همکاران ، 2002 ؛ Young و همکاران ، 2002). در واقع ، ناهنجاری های برنامه ریزی شده ژنتیکی متابولیسم سوخت قلب در مدل های موش باعث تحریک کاردیومیوپاتی می شود (Carley et al.، 2014؛ Neubauer، 2007). در شرایط فیزیولوژیکی ، قلب های طبیعی بدن کتونی را متناسب با زایمان آنها اکسیداسیون می کنند ، این امر به قیمت اسیدهای چرب و اکسیداسیون گلوکز انجام می شود و میوکارد بیشترین مصرف کننده بدن کتون در واحد جرم است (BING، 1954؛ Crawford et al.، 2009؛ GARLAND و همکاران) . ، 1962 ؛ Hasselbaink و همکاران ، 2003 ؛ جفری و همکاران ، 1995 ؛ پلتیر و همکاران ، 2007 ؛ Tardif و همکاران ، 2001 ؛ یان و همکاران ، 2009). در مقایسه با اکسیداسیون اسیدهای چرب ، اجسام کتون از نظر انرژی کارآمدتر هستند و انرژی بیشتری را برای سنتز ATP به ازای هر مولکول اکسیژن سرمایه گذاری شده (نسبت P / O) تولید می کنند (Kashiwaya و همکاران ، 2010 ؛ Sato و همکاران ، 1995 ؛ Veech ، 2004) . اکسیداسیون بدن کتون همچنین به طور بالقوه انرژی بیشتری نسبت به فاو تولید می کند ، و اوبی کوینون را اکسید می کند ، که باعث کاهش دهانه اکسایش اکسیداسیون در زنجیره انتقال الکترون می شود و انرژی بیشتری را برای سنتز ATP فراهم می کند (Sato et al.، 1995؛ Veech، 2004). اکسیداسیون اجسام کتون همچنین ممکن است تولید ROS و در نتیجه استرس اکسیداتیو را کاهش دهد (Veech ، 2004).
مطالعات اولیه مداخله ای و مشاهده ای نشان می دهد که نقش بالقوه ای در بدن کتون در قلب وجود دارد. در زمینه آسیبهای ایسکمی / رپرفیوژن تجربی، اعضای بدن کتون، توانایی بالقوه محافظت در برابر قلبتان را دارند (الزید و همکاران، 2007؛ وانگ و همکاران، 2008)، احتمالا به دلیل افزایش فراوانی میتوكندری در قلب یا تنظیم مقابله با فسفوریلاسیون اكسیداتیک حیاتی واسطه ها (Snorek و همکاران، 2012؛ Zou و همکاران، 2002). مطالعات اخیر نشان می دهد که استفاده از بدن کتون در دل شکستگی موش ها (Aubert و همکاران، 2016) و انسان ها (Bedi et al.، 2016)، از مشاهدات قبلی در انسان پشتیبانی می کند (BING، 1954؛ Fukao et al.، 2000؛ Janardhan و همکاران، 2011، Longo و همکاران، 2004، رودولف و Schinz، 1973، Tildon و Cornblath، 1972). غلظت کتون در گردش خون در بیماران مبتلا به نارسایی قلب افزایش می یابد، به طور مستقیم با فشار پر شدن، مشاهدات که مکانیزم و اهمیت آن هنوز معلوم نیست (Kupari و همکاران، 1995، Lommi و همکاران، 1996، Lommi و همکاران، 1997، Neely و همکاران .، 1972)، اما موش هایی با کمبود SCOT انتخابی در کراتومیوسیت ها، در بهبود واکنش های بطنی پاتولوژیک بطن ها و واکنش های ROS در پاسخ به آسیب های اضطراب تحت فشار جراحی (Schugar و همکاران، 2014) افزایش یافته است.
مشاهدات جالب توجه اخیر در دیابت درمان یک پیوند بالقوه بین متابولیسم کتون های قلب و ساختار مجدد بطنی پاتولوژیک را نشان داد (شکل 5). غلظت 2 (SGLT2i)، پروتئین سولفوریک پروتئین سولفوریک / گلوکز (SGLT2016i) باعث افزایش غلظت کتون بدن در انسان (Ferrannini و همکاران، 2015a، Inagaki و همکاران، 2014) و موش (Suzuki و همکاران، 2014) از طریق افزایش کتوژنز کبدی (Ferrannini و همکاران، 2016، Ferrannini و همکاران، 2015a، Katz و Leiter، 2015، Mudaliar و همکاران، 2016). به طور قابل توجهی، حداقل یکی از این عوامل باعث کاهش زمان بستری شدن HF (به عنوان مثال در آزمایش EMPA-REG) و بهبود مرگ و میر قلبی عروقی (Fitchett و همکاران، 2016؛ Sonesson و همکاران، 2016؛ Wu و همکاران، 2015a ؛ Zinman و همکاران، 2). در حالی که مکانیسم های راننده پس از نتایج سودمند HF برای اتصال SGLT2016i به طور فعال مورد بحث قرار می گیرد، مزایای زنده ماندن احتمالا چند فاکتوریل است، در آینده شامل کتوز، اما همچنین اثرات سالم بر وزن، فشار خون، گلوکز و اسید اوریک، سختی شریان، سیستم عصبی سمپاتیک، اسمزی دیورتیک / کاهش حجم پلاسما و افزایش هماتوکریت (راز و کان، 2016، والون و تامسون، 2016). در کنار هم، مفهوم افزایش کتونمی درمانی در بیماران HF یا افرادی که در معرض خطر ابتلا به HF قرار دارند، همچنان بحث برانگیز است، اما تحت تحقیق فعال در مطالعات پیش از بالینی و بالینی قرار دارد (Ferrannini et al.، 2016b؛ Kolwicz et al. 2016؛ Lopaschuk و Verma؛ 2016؛ Mudaliar و همکاران، 2016؛ Taegtmeyer، XNUMX).
�
بدن کتون در زیست شناسی سرطان
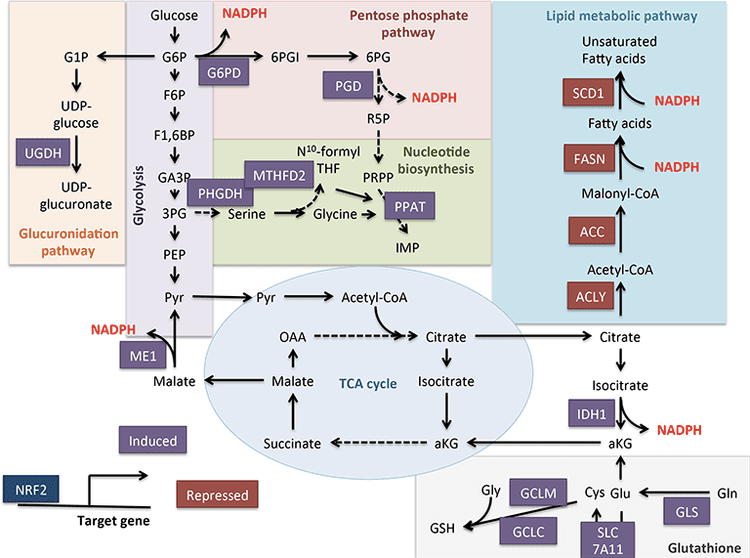
ارتباط بین بدن کتون و سرطان به سرعت در حال ظهور است، اما مطالعه در هر دو مدل حیوانی و انسان نتیجه های متنوعی به دست آورده است. از آنجایی که متابولیسم کتون پویا و حالت مغذی پاسخگو است، لذت بردن از پیوند زیستی با سرطان به دلیل پتانسیل درمان های تغذیه با هدایت دقیق است. سلول های سرطانی تحت برنامه ریزی متابولیکی برای حفظ سریع تکثیر و رشد سلولی (DeNicola و Cantley، 2015، Pavlova و Thompson، 2016) قرار می گیرند. اثر Warburg کلاسیک در متابولیسم سلول های سرطانی ناشی از نقش غالب گلوکز و تخمیر اسید لاکتیک برای انتقال انرژی و جبران وابستگی کمتر به فسفوریلاسیون اکسیداتیو و تنفس میتوکندری محدود است (De Feyter et al.، 2016؛ Grabacka et al.، 2016؛ Kang و همکاران، 2015؛ پوف و همکاران، 2014؛ Shukla و همکاران، 2014). کربن گلوکز عمدتا از طریق گلیکولیز، مسیر پنتوز فسفات و لیپوژنز هدایت می شود که به طور همزمان واسنجی لازم برای گسترش زیست توده تومور را فراهم می کند (Grabacka و همکاران، 2016، Shukla و همکاران، 2014، Yoshii و همکاران، 2015). انطباق سلول های سرطانی با محرومیت گلوکز از طریق توانایی استفاده از منابع جایگزین سوخت، از جمله استات، گلوتامین و آسپارتات می باشد (Jaworski et al.، 2016؛ Sullivan et al.، 2015). برای مثال، دسترسی محدود به پریووت نشان دهنده توانایی سلول های سرطانی برای تبدیل گلوتامین به استیل کولا بوسیله کربوکسیلینگ، حفظ نیازهای انرژی و آنابولیک است (یانگ و همکاران، 2014). سازگاری جالب از سلول های سرطانی، استفاده از استات به عنوان یک سوخت است (Comerford و همکاران، 2014، Jaworski و همکاران، 2016، Mashimo و همکاران، 2014، رایت و سیمون، 2016، Yoshii و همکاران، 2015). استات نیز یک بستر برای لیپوژنز است که برای تکثیر سلول های تومور حیاتی است و افزایش این کانال لیپوژنیک با بقای کوتاه مدت بیمار و افزایش تومور بیشتر مرتبط است (Comerford و همکاران، 2014، Mashimo و همکاران، 2014، Yoshii et al .، 2015).
سلولهای غیرسرطانی در طی محرومیت از گلوکز ، به راحتی منبع انرژی خود را از گلوکز به بدن کتون تغییر می دهند. این انعطاف پذیری ممکن است در میان انواع سلولهای سرطانی متغیر باشد ، اما در داخل بدن تومورهای مغز کاشته شده اکسید شده [2,4،13-2C2016] -؟ OHB تا حدی مانند بافت اطراف مغز (De Feyter et al.، 2010). اثر معکوس اثر واربورگ یا دو متابولیسم تومور محفظه ای این فرضیه را ایجاد می کند که سلول های سرطانی باعث تولید OHH در فیبروبلاست های مجاور می شوند و نیازهای انرژی سلول تومور را تأمین می کنند (بونوچلی و دیگران ، 2012 ؛ مارتینز-اوتسچورن و همکاران ، 1) . در کبد ، تغییر سلولهای کبدی از کتوژنز به اکسیداسیون کتون در سلولهای سرطان سلولهای کبدی (هپاتوم) با فعال سازی فعالیتهای BDH1989 و SCOT مشاهده شده در دو رده سلول هپاتوم سازگار است (ژانگ و همکاران ، 1). در واقع ، سلولهای کبدی OXCT1 و BDH2016 را بیان می کنند و کتونها را اکسید می کنند ، اما فقط در صورت گرسنگی سرم (هوانگ و همکاران ، 2). از طرف دیگر ، کتوژنز سلول توموری نیز پیشنهاد شده است. جابجایی های دینامیکی در بیان ژن کتوژنیک در طی تغییر شکل سرطانی اپیتلیوم روده بزرگ ، یک نوع سلول که به طور معمول HMGCS2 را بیان می کند ، به نمایش گذاشته می شود و گزارش اخیر نشان می دهد که HMGCS2006 ممکن است یک نشانگر پیش آگهی پیش آگهی ضعیف در سرطان سلول های روده بزرگ و سنگفرشی باشد (Camarero و همکاران ، 2016 ؛ چن و همکاران ، 2). اینکه آیا این ارتباط به کتوژنز نیاز دارد یا شامل آن می شود ، یا عملکرد مهتابی HMGCS2016 را مشخص می کند. برعکس ، تولید OHB توسط سلولهای ملانوم و گلیوبلاستوما ، تحریک شده توسط PPAR؟ آگونیست fenofibrate ، با توقف رشد همراه بود (Grabacka و همکاران ، 2). برای مشخص کردن نقش بیان HMGCSXNUMX / SCOT ، کتوژنز و اکسیداسیون کتون در سلولهای سرطانی ، مطالعات بیشتری لازم است.
فراتر از حوزه متابولیسم سوخت ، اخیراً کتونها از طریق مکانیزم سیگنالینگ در زیست سلولهای سرطانی نقش دارند. تجزیه و تحلیل ملانومای BRAF-V600E + نشان داد که القای HMGCL وابسته به OCT1 به روشی وابسته به BRAF سرطان زا است (کانگ و همکاران ، 2015). افزایش HMGCL با غلظت بالاتر سلولی AcAc در ارتباط بود ، که به نوبه خود تعامل BRAFV600E-MEK1 را افزایش می دهد ، با تقویت سیگنالینگ MEK-ERK در یک حلقه خوراکی که باعث تکثیر و رشد سلول های توموری می شود. این مشاهدات سوال جالب توجهی از کتوژنیز خارج کبدی احتمالی را ایجاد می کند که سپس از یک مکانیسم سیگنالینگ پشتیبانی می کند (همچنین به OHB به عنوان یک واسطه سیگنالینگ و اختلافات در کتوژنز خارج کبدی مراجعه کنید). همچنین مهم است که اثرات مستقل AcAc ، d-؟ OHB و l-؟ OHB بر متابولیسم سرطان در نظر گرفته شود ، و هنگام بررسی HMGCL ، کاتابولیسم لوسین نیز ممکن است از بین برود.
اثرات رژیم های کتوژنیک (همچنین به درمان درمانی رژیم کتوژنیک و اجسام کتون برونزا مراجعه کنید) در مدل های حیوانی سرطانی متنوع است (De Feyter et al.، 2016؛ Klement et al.، 2016؛ Meidenbauer et al.، 2015؛ Poff et al. . ، 2014 ؛ سیفرید و همکاران ، 2011 ؛ شوکلا و همکاران ، 2014). در حالی که ارتباطات اپیدمیولوژیک بین چاقی ، سرطان و رژیم های کتوژنیک مورد بحث است (Liskiewicz و همکاران ، 2016 ؛ رایت و سیمون ، 2016) ، یک متاآنالیز با استفاده از رژیم های کتوژنیک در مدل های حیوانی و در مطالعات انسانی تأثیر سلامتی بر بقا را نشان داد ، با مزایای آینده نگر به بزرگی کتوز ، زمان شروع رژیم غذایی و محل تومور مرتبط است (کلمنت و همکاران ، 2016 ؛ وولف و همکاران ، 2016). درمان سلولهای سرطانی پانکراس با اجسام کتون (d-؟ OHB یا AcAc) رشد ، تکثیر و گلیکولیز و رژیم کتوژنیک (81٪ کیلو کالری چربی ، 18٪ پروتئین ، 1٪ کربوهیدرات) را کاهش می دهد ، از نظر وزن بدن تومور ، گلیسمی و افزایش عضله و وزن بدن در حیوانات مبتلا به سرطان کاشته شده (شوکلا و همکاران ، 2014). نتایج مشابهی با استفاده از مدل سلول گلیوبلاستوما متاستاتیک در موشهایی که مکمل کتون در رژیم غذایی دریافت کردند مشاهده شد (پوف و همکاران ، 2014). برعکس ، یک رژیم کتوژنیک (91٪ کیلو کالری چربی ، 9٪ پروتئین) باعث افزایش غلظت OHB در گردش خون و کاهش گلیسمی می شود ، اما هیچ تأثیری بر حجم تومور یا مدت زنده ماندن در موشهای صحرایی وجود ندارد (De Feyter و همکاران ، 2016). یک شاخص گلوکز کتون به عنوان یک شاخص بالینی ارائه شده است که باعث بهبود مدیریت متابولیسم درمان سرطان مغز ناشی از رژیم کتوژنیک در انسان و موش می شود (Meidenbauer et al.، 2015). روی هم رفته ، نقش متابولیسم بدن کتون و اجسام کتونی در زیست شناسی سرطان دلهره آور است زیرا هر یک از آنها گزینه های درمانی قابل حل است ، اما جنبه های اساسی باید توضیح داده شوند ، با تأثیرات واضح از ماتریس متغیرها ، از جمله (من) تفاوت بین کتون برون زا بدن در مقابل رژیم کتوژنیک ، (ii) نوع سلول سرطانی ، چندشکلی ژنومی ، درجه و مرحله ؛ و (III) زمان و مدت زمان قرار گرفتن در معرض حالت کتوتیک.
کتوژنیزاسیون بوسیله تجزیه اسیدهای چرب و اسیدهای آمینه اتیلن توسط اجزای کتون ایجاد می شود. این فرایند بیوشیمی انرژی را به ارگان های مختلف، به ویژه مغز، در شرایط ناشتا به عنوان پاسخ به عدم دسترسی به قند خون، انرژی می دهد. بدن کتون ها عمدتا در میتوکندری سلول های کبدی تولید می شود. در حالی که سلول های دیگر قادر به انجام کتونی هستند، در سلول های کبدی کارایی ندارند. از آنجا که کتوژنز در میتوکندری اتفاق می افتد، فرآیندهای آن به طور مستقل تنظیم می شود. دکتر الکس جیمنز DC، CCST Insight
کاربرد درمانی رژیم کتونی و بدن کتون خارجی
کاربردهای رژیم های کتوژنیک و اجسام کتون به عنوان ابزار درمانی در زمینه های غیر سرطانی از جمله چاقی و NAFLD / NASH نیز به وجود آمده است (براونینگ و همکاران ، 2011 ؛ فاستر و همکاران ، 2010 ؛ شوگر و کرافورد ، 2012). نارسایی قلبی (Huynh، 2016؛ Kolwicz et al.، 2016؛ Taegtmeyer، 2016) ؛ بیماری عصبی و تخریب عصبی (مارتین و همکاران ، 2016 ؛ مک نالی و هارتمن ، 2012 ؛ رو ، 2015 ؛ روگاوسکی و همکاران ، 2016 ؛ یانگ و چنگ ، 2010 ؛ یائو و همکاران ، 2011) ؛ خطاهای ذاتی متابولیسم (Scholl-B rgi و همکاران ، 2015) ؛ و عملکرد ورزشی (کاکس و همکاران ، 2016). اثر رژیم های کتوژنیک به ویژه در درمان تشنج صرع ، به ویژه در بیماران مقاوم به دارو ، قدردانی شده است. اکثر مطالعات رژیم های کتوژنیک را در بیماران کودکان ارزیابی کرده اند و نشان می دهد که تا 50 reduction کاهش فراوانی تشنج پس از 3 ماه ، با بهبود اثر در سندرم های انتخاب شده (Wu et al.، 2016b). این تجربه در صرع بزرگسالان محدودتر است ، اما کاهش مشابه آن ، با پاسخ بهتر در بیماران مبتلا به صرع عمومی علامت دار ، مشهود است (نی و همکاران ، 2014). مکانیسم های ضد تشنج زمینه ای نامشخص است ، اگرچه فرضیه های فرضی شامل کاهش استفاده از گلوکز / گلیکولیز ، حمل و نقل مجدد برنامه ریزی شده گلوتامات ، تأثیر غیرمستقیم بر کانال پتاسیم حساس به ATP یا گیرنده آدنوزین A1 ، تغییر بیان ایزوفرم کانال سدیم یا تأثیر بر هورمون های در گردش از جمله لپتین ( Lambrechts et al.، 2016؛ Lin et al.، 2017؛ Lutas and Yellen، 2013). هنوز مشخص نیست که آیا اثر ضد تشنج عمدتاً به بدن کتون نسبت داده می شود یا به دلیل عواقب متابولیکی آبشارهای رژیم های کم کربوهیدرات است. با این حال ، به نظر می رسد استرهای کتون (در زیر را ببینید) آستانه تشنج را در مدل های حیوانی تشنج های تحریک شده بالا می برند (سیارلون و همکاران ، 2016 ؛ داگوستینو و همکاران ، 2013 ؛ ویگیانو و همکاران ، 2015).
رژیم های کم کربوهیدرات سبک اتکینز و کتوژنیک اغلب غیرمعمول هستند و می توانند باعث یبوست، هیپوریکمی، هیپوکلسمی، هیپومنیزمی، منجر به نفروتیلاسیون، کتواسیدوز، ایجاد هیپرگلیسمی و افزایش کلسترول تجمع یافته و غلظت اسید چرب آزاد شوند (Bisschop et al.، 2001 ؛ Kossoff و Hartman، 2012؛ Kwiterovich و همکاران، 2003؛ Suzuki و همکاران، 2002). به همین علت پیوستگی درازمدت چالش هایی را ایجاد می کند. مطالعات جوندگان معمولا از توزیع مغذی متمایز استفاده می کنند (94٪ کیلوگرم چربی، 1٪ کربوهیدرات کیلوکالری، پروتئین KNK 5 پروتئین، Bio-Serv F3666)، که باعث کتوز قوی می شود. با این حال، افزایش محتوای پروتئین، حتی به 10٪ kcal به طور قابل ملاحظه ای کتوز را کاهش می دهد، و محدودیت پروتئین KKAL 5 اثرات متابولیکی و فیزیولوژیکی را مخدوش می کند. این فرمول رژیم غذایی نیز کولین تخلیه شده است، یکی دیگر از متغیرهایی است که حساسیت به آسیب های کبدی و حتی کتوژنز را تحت تاثیر قرار می دهد (Garbow و همکاران، 2011، جورنایواز و همکاران، 2010، کندی و همکاران، 2007، Pissios و همکاران، 2013، Schugar و غیره، 2013). اثرات مصرف طولانی مدت رژیم های کتوژنیک در موش هنوز به طور کامل تعریف نشده است، اما مطالعات اخیر در موش ها نشان می دهد که بقای طبیعی و عدم نشانگر آسیب کبدی در موش ها در طول رژیم های غذایی کتوژنیک، هرچند متابولیسم آمینو اسید، هزینه انرژی و سیگنالینگ انسولین به طور قابل توجهی دوباره برنامه ریزی شده بودند (Douris et al.، 2015).
مکانیسم های افزایش کتوز از طریق مکانیسم های جایگزین برای رژیم های کتوژنیک شامل استفاده از پیش سازهای کتون در دسترس می باشد. اعمال بدن کتون خارجی ممکن است یک حالت فیزیولوژیک منحصر به فرد ایجاد کند که در فیزیولوژی طبیعی مواجه نشود زیرا غلظت گلوکز و غلظت انسولین نسبتا نرمال است، در حالی که سلول ها ممکن است جذب و مصرف گلوکز را از بین ببرند. بدن کتون ها نیمه عمر کوتاهی دارند و بلع یا تزریق نمک سدیم؟ OHB برای دستیابی به کتوز درمانی باعث تحریک بار سدیم ناخوشایند می شود. R / S-1,3،1992-butanediol یک الکل غیر سمی است که به راحتی در کبد اکسید می شود و باعث تولید d / l-؟ OHB می شود (Desrochers et al.، XNUMX). در زمینه های تجربی مجزا ، این دوز به مدت هفت هفته به طور روزانه بر روی موش یا موش تجویز می شود و در طی 5 ساعت پس از تجویز غلظت های OHB تا 2 میلی مولار در گردش است که حداقل برای 3 ساعت اضافی (D ') پایدار است. آگوستینو و دیگران ، 2013). سرکوب بخشی از مصرف غذا در جوندگان دیده شده است که به R / S-1,3-Butanediol (Carpenter and Grossman، 1983) داده شده است. علاوه بر این ، سه استر کتون متمایز از نظر شیمیایی (KE) ، (من) مونوستر R-1,3،3-بوتاندیول و d-؟ OHB (R-1,3-هیدروکسی بوتیل R-؟ OHB). (ii) گلیسیریل-تریس-؟ OHB ؛ و (iii) R ، S-1997،2012-butanediol acetoacetate diester نیز به طور گسترده مورد مطالعه قرار گرفته است (Brunengraber، 2012؛ Clarke et al.، 1995a؛ Clarke et al.، 1995b؛ Desrochers et al.، 2010a؛ Desrochers et al. . ، XNUMXb ؛ Kashiwaya و همکاران ، XNUMX). یک مزیت ذاتی مورد اول این است که به دنبال هیدرولیز استراز در روده یا کبد ، 2 مول d-OHHB فیزیولوژیکی در هر مول KE تولید می شود. ایمنی ، فارماکوکینتیک و تحمل به طور گسترده در انسانهایی که R-3-hydroxybutyl R-؟ OHB مصرف می کنند ، در دوزهای حداکثر 714 میلی گرم در کیلوگرم ، و غلظت های d-؟ OHB در گردش را تا 6 میلی متر ارائه می دهد (کلارک و همکاران ، 2012a ؛ کوکس و همکاران ، 2016 ؛ کمپر و همکاران ، 2015 ؛ شیووا و همکاران ، 2016). در جوندگان، این KE کاهش مصرف کالری و کلسترول کلسترول را افزایش می دهد، بافت چربی قهوه ای را تحریک می کند و مقاومت به انسولین را بهبود می بخشد (Kashiwaya et al.، 2010؛ Kemper et al.، 2015؛ Veech، 2013). یافته های اخیر نشان می دهد که در حین ورزش در ورزشکاران آموزش دیده ، بلع R-3-hydroxybutyl R-؟ OHB باعث کاهش گلیکولیز عضله اسکلتی و غلظت لاکتات پلاسما ، افزایش اکسیداسیون تری اسیل گلیسرول عضلانی و حفظ محتوای گلیکوژن عضله ، حتی در هنگام مصرف همزمان کربوهیدرات ترشح انسولین () کاکس و همکاران ، 2016). توسعه بیشتر این نتایج جالبی لازم است، زیرا بهبود عملکرد استقامتی عمدتا توسط پاسخ قوی به KE در افراد 2 / 8 هدایت می شود. با این وجود، این نتایج از مطالعات کلاسیک حمایت می کند که نشان دهنده اولویت اکسیداسیون کتون بر روی دیگر مواد (GARLAND و همکاران، 1962، Hasselbaink و همکاران، 2003، Stanley و همکاران، 2003، Valente-Silva و همکاران، 2015) از جمله در حین ورزش، و این که ورزشکاران آموزش دیده ممکن است برای استفاده از کتون ها به کار گرفته شوند (جانسون و همکاران، 1969a، جانسون و والنتن، 1972، Winder و همکاران، 1974، Winder و همکاران، 1975). در نهایت، مکانیسم هایی که ممکن است عملکرد ورزشی بهبود یافته را با در نظر گرفتن کالری یکسانی (که به صورت توزیع بین مواد مغذی توزیع شده است) و میزان مصرف اکسیژن مساوی باقی بماند، هنوز تعیین می شود.
چشم انداز آینده
مشاهدات اخیر که به عنوان یک مسیر سرریز که قادر به جمع آوری انتشار سمی از احتراق چربی در کشورهای محدود شده با کربوهیدرات است ، انگ زده می شود ، این مفهوم را پشتیبانی می کند که متابولیسم بدن کتون حتی در حالت های پر از کربوهیدرات نیز نقش های نجات دهنده ایفا می کند ، فرضیه در حالی که رویکردهای تغذیه ای و دارویی آسان برای دستکاری متابولیسم کتون ، آن را به یک هدف درمانی جذاب تبدیل می کند ، اما آزمایشات محتاطانه ای در هر دو آزمایشگاه تحقیق اولیه و ترجمه باقی مانده است. نیازهای برآورده نشده در زمینه های تعیین نقش استفاده از متابولیسم کتون در نارسایی قلبی ، چاقی ، NAFLD / NASH ، دیابت نوع 2 و سرطان ظاهر شده است. دامنه و تأثیر نقشهای "غیر متعارف" سیگنالینگ اجسام کتون ، از جمله تنظیم PTM ها که احتمالاً به مسیرهای متابولیکی و سیگنالینگ تغذیه می شوند و به جلو هدایت می شوند ، به کاوش عمیق تری نیاز دارند. سرانجام ، کتوژنز خارج کبدی می تواند مکانیسم های سیگنالینگ پاراکرین و اتوکرین جذاب و فرصت هایی را برای تأثیر بر متابولیسم در سیستم عصبی و تومورها برای دستیابی به اهداف درمانی باز کند.
در نتیجه ، بدن کتون توسط کبد ایجاد می شود تا وقتی که گلوکز کافی در بدن انسان موجود نیست ، به عنوان منبع انرژی استفاده شود. کتوژنز زمانی اتفاق می افتد که سطح گلوکز در خون کم باشد ، به ویژه پس از اتمام سایر ذخایر کربوهیدرات سلولی. هدف مقاله بالا بحث در مورد نقشهای چند بعدی بدن کتون در متابولیسم سوخت ، سیگنالینگ و درمانها بود. دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
درد پشتاین یکی از مهمترین دلایل معلولیت و روزهای از دست رفته در کار در سراسر جهان است. کمردرد دومین دلیل شایع مراجعه به مطب توسط پزشکان است که بیشتر از عفونت های دستگاه تنفسی فوقانی است. تقریباً 80 درصد مردم حداقل یک بار در طول زندگی خود درد کمر را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده است که از استخوان ها ، مفاصل ، رباط ها و ماهیچه ها در میان سایر بافت های نرم تشکیل شده است. آسیب ها و / یا شرایط وخیم ، ماننددیسک های فتق دیسک، می تواند در نهایت به علائم کمردرد منجر شود. آسیب های ورزشی یا صدمات ناشی از تصادفات اتومبیل اغلب شایعترین علت کمر درد است ، با این حال ، گاهی اوقات ساده ترین حرکات می تواند نتایج دردناکی داشته باشد. خوشبختانه ، گزینه های درمانی جایگزین مانند مراقبت های کایروپراکتیک ، می توانند با استفاده از تنظیمات ستون فقرات و دستکاری های دستی ، درد کمر را کاهش دهند و در نهایت تسکین درد را بهبود بخشند.
La 2 عامل مربوط به 2 هسته ای سیگنالینگ، که به بهترین شکل به نام Nrf2 شناخته می شود، مکانیسم محافظتی است که به عنوان یک رگولاتور اصلی پاسخ آنتی اکسیدانی بدن انسان عمل می کند. Nrf2 سطوح استرس اکسیداتیو را درون سلول ها می شناسد و مکانیزم های آنتی اکسیدانی محافظ را باعث می شود. در حالی که Activation Nrf2 می تواند مزایای بسیاری داشته باشد، "overexpression" Nrf2 می تواند خطرات متعددی داشته باشد.
به نظر می رسد که درجه متعادل NRF2 در جلوگیری از توسعه کلی بیماری های مختلف علاوه بر بهبود کلی این مسائل بهداشتی ضروری است. با این حال، NRF2 همچنین می تواند عوارض ایجاد کند. علت اصلی NRF2 "بیش از حد بیان" به علت جهش ژنتیکی یا مداوم در معرض استرس شیمیایی یا استرس اکسیداتیو است. در زیر، ما در مورد مزایای بیش از حد بیان Nrf2 بحث و نشان دادن مکانیسم عمل آن در بدن انسان است.
سرطان
مطالعات تحقیقاتی نشان داد موشهایی که NRF2 را بیان نمی کنند در پاسخ به تحریکات فیزیکی و شیمیایی تمایل بیشتری به ابتلا به سرطان دارند. با این حال ، مطالعات تحقیقاتی مشابه نشان داد که فعال شدن بیش از حد NRF2 یا حتی غیرفعال سازی KEAP1 می تواند منجر به تشدید برخی سرطان ها شود ، به خصوص اگر این مسیرها قطع شده باشد. بیش فعال NRF2 می تواند از طریق سیگار کشیدن رخ دهد ، جایی که اعتقاد بر این است فعال شدن مداوم NRF2 دلیل سرطان ریه در افراد سیگاری است. بیان بیش از حد Nrf2 ممکن است باعث شود سلول های سرطانی خود تخریب نشوند ، در حالی که فعال شدن NRF2 متناوب می تواند از تحریک القای سم به سلول های سرطانی جلوگیری کند.
بعلاوه ، از آنجا که بیان بیش از حد NRF2 توانایی آنتی اکسیدانی بدن انسان را برای عملکرد فراتر از هموستاز ردوکس افزایش می دهد ، این تقسیم سلولی را افزایش می دهد و الگوی غیرطبیعی DNA و متیلاسیون هیستون را ایجاد می کند. در نهایت این می تواند شیمی درمانی و رادیوتراپی را در برابر سرطان کمتر م lessثر سازد. بنابراین ، محدود کردن فعال سازی NRF2 با موادی مانند DIM ، لوتئولین ، زی کائو یا سالینومایسین می تواند برای بیماران مبتلا به سرطان ایده آل باشد اگرچه نباید بیش از حد فعال شدن Nrf2 را تنها علت سرطان دانست. کمبود مواد مغذی می تواند ژن ها را تحت تأثیر قرار دهد ، از جمله NRF2. این ممکن است یکی از راههای چگونگی کمبودها در ایجاد تومورها باشد.
کبد
Overactivation از Nrf2، همچنین می تواند بر عملکرد ارگان های خاص بدن انسان تأثیر بگذارد. فراتر از بیان NRF2 در نهایت می تواند تولید فاکتور رشد انسولین 1 یا IGF-1 را از کبد، که برای بازسازی کبد ضروری است، مسدود کند.
قلب
در حالی که بیش از حد بیان حاد از Nrf2 ممکن است منافع خود را داشته باشد، بیش از حد Expression of NRF2 مداوم می تواند اثرات مضر طولانی مدت بر روی قلب، مانند cardiomyopathy. بیان NRF2 می تواند از طریق افزایش سطح کلسترول و یا فعال شدن HO-1 افزایش یابد. اعتقاد بر این است که چرا سطح بالای کلسترول مزمن ممکن است سبب مشکلات سلامت قلبی و عروقی شود.
ویتیلیگو
بیش از حد بیان NRF2 نیز نشان داده شده است که مانع از توانایی رسوب زدن در ویتیلیگو می شود، زیرا ممکن است فعالیت Tyrosinase یا TYR را مختل کند که برای پپ منگنه شدن از طریق ملانین زایی ضروری است. مطالعات تحقیقاتی نشان داده است که این فرایند ممکن است یکی از دلایل اصلی این باشد که چرا افراد مبتلا به ویتیلیگو به طور موثر به عنوان افراد بدون ویتیلیگو فعال Nrf2 فعال می کنند.
چرا NRF2 ممکن است به درستی کار نکند
هورمون
NRF2 باید از نظر هورمونی فعال شود تا بتواند از مزایای آن بهره ببرد. به عبارت دیگر ، Nrf2 نباید هر دقیقه یا هر روز باعث تحریک شود ، بنابراین ، ایده خوبی است که از آن استراحت کنید ، به عنوان مثال ، 5 روز در 5 روز تعطیل یا هر روز در میان. NRF2 همچنین باید یک آستانه خاص را برای تحریک پاسخ هورمونی خود به دست آورد ، جایی که یک عامل فشار آور کوچک برای تحریک آن کافی نیست.
اکسیداسیون DJ-1
پروتئین deglycase DJ-1، یا فقط DJ-1، همچنین پروتئین بیماری پارکینسون نامیده می شود، یا PARK7، تنظیم کننده اصلی و آشکارساز وضعیت بازسازی رادیواکتیو در بدن انسان است. DJ-1 برای تنظیم اینکه چه مدت NRF2 می تواند عملکرد آن را انجام دهد و پاسخ آنتی اکسیدانی تولید کند، ضروری است. در مواردی که DJ-1 بیش از حد اکسید شده باشد، سلول ها پروتکل DJ-1 را کمتر قابل دسترسی خواهند کرد.
این فرآیند باعث می شود تا فعال شدن NRF2 خیلی سریع به پایان برسد زیرا DJ-1 برای حفظ سطوح متوازن NRF2 و جلوگیری از آن در سلول شکسته است. در صورتی که پروتئین DJ-1 غیرممکن یا بیش از حد اکسید شده باشد، بیان NRF2 احتمالا حداقل خواهد بود، حتی با استفاده از DIM یا جایگزین NRF2. بیان DJ-1 ضروری است برای بازگرداندن عمل NRF2 مختل شود.
بیماری مزمن
اگر به بیماری مزمن از جمله CIRS ، عفونت های مزمن / دیس بیوزیس / SIBO یا تجمع فلزات سنگین مانند جیوه و یا کانال کانال ریشه مبتلا هستید ، این می تواند سیستم NRF2 و سم زدایی فاز دو را مختل کند. به جای اینکه استرس اکسیداتیو NRF2 را به یک آنتی اکسیدان تبدیل کند ، NRF2 تحریک نخواهد شد و استرس اکسیداتیو می تواند در سلول باقی بماند و باعث آسیب شود ، به این معنی که هیچ پاسخ آنتی اکسیدانی وجود ندارد. این دلیل قابل توجهی است که بسیاری از افراد مبتلا به CIRS دارای چندین حساسیت هستند و به عوامل بی شماری دست می یابند. برخی از افراد معتقدند كه ممكن است دارای پاسخ هرگز باشند ، با این حال ، این واکنش فقط می تواند به سلولهای دورتر آسیب برساند.
با این حال ، درمان بیماری مزمن به کبد اجازه می دهد تا سموم را در صفرا تخلیه کند و به تدریج پاسخ هورمونی فعال شدن NRF2 را ایجاد کند. اگر صفرا سمی باقی بماند و از بدن انسان دفع نشود ، استرس اکسیداتیو NRF2 را دوباره فعال می کند و باعث می شود پس از جذب مجدد از دستگاه گوارش یا دستگاه گوارش ، احساس بدتری داشته باشید. به عنوان مثال ، اوکراتوکسین A ممکن است NRF2 را مسدود کند. گذشته از درمان مشکل ، مهار کننده های هیستون دی استیلاز می توانند واکنش اکسیداتیو را از طریق تعدادی از عواملی که باعث فعال شدن NRF2 می شوند ، مسدود کنند ، اما همچنین ممکن است از تحریک غیر طبیعی NRF2 جلوگیری کند ، که ممکن است در نهایت به هدف خود منجر نشود.
اختلال در تنظیم روغن ماهی
کولینرژیک ها مواد هستند که باعث افزایش استیل کولین، یا ACh و کولین در مغز از طریق افزایش ACh، به ویژه هنگامی که مهار تجزیه ACh. بیماران مبتلا به CIRS اغلب دارای اختلال در تنظیم مقادیر استیل کولین در بدن انسان، به ویژه در مغز هستند. روغن ماهی باعث می شود NRF2، فعال سازی مکانیزم آنتی اکسیدانی محافظ در داخل سلول ها.
افراد مبتلا به بیماری های مزمن ممکن است در اثر استرس شناختی و مسمومیت با استیل کولین ، از تجمع ارگانوفسفات ، که باعث ایجاد روغن ماهی در بدن انسان می شود ، مشکل داشته باشند. کمبود کولین علاوه بر این باعث فعال شدن NRF2 می شود. گنجاندن کولین در رژیم غذایی ، (پلی فنول ، تخم مرغ و غیره) می تواند به تقویت اثرات تنظیم نظم کولینرژیک کمک کند.
چه چیزی NRF2 را کاهش می دهد؟
کاهش بیان بیش از حد NRF2 برای افرادی که مبتلا به سرطان هستند بهتر است، اگر چه ممکن است برای بسیاری از مسائل دیگر سلامت مفید باشد.
رژیم غذایی، مکمل ها و داروهای رایج:
آپی ژنین (دوزهای بالاتر)
بروسا جاوینیکا
شاه بلوط
EGCG (دوزهای بالا NRF2 را افزایش می دهد)
Fenugreek (Trigonelline)
هیبا (Hinokitiol /؟ -thujaplicin)
رژیم غذایی نمک زیاد
لوتئولین (کرفس، فلفل، جعفری، برگ پریلا و چای بابونه - دوزهای بالاتر ممکن است NRF2 - 40 mg / kg لووتولین را سه بار در هفته افزایش دهد)
متفورمین (مصرف مزمن)
N-Acetyl-L-Cysteine (NAC، با مسدود کردن پاسخ اکسیداتیو، ESP در دوزهای بالا)
پوست پرتقال (دارای فلاونوئیدهای پلیتوکسیل شده)
Quercetin (دوزهای بالاتر ممکن است NRF2 - 50 mg / kg / d quercetin را افزایش دهد)
سالینوماسین (دارو)
رتینول (اسید رتینوئیک تمام ترانس)
ویتامین C در ترکیب با Quercetin
Zi Cao (بنفش Gromwel دارای Shikonin / Alkannin)
مسیرها و سایر موارد:
Bach1
شرط می بندم
بیوفیلم ها
بروساتول
کمپتوتیسین
DNMT
DPP-23
EZH2
سیگنال گیرنده گلوکوکورتیکوئیدی (دگزامتازون و بتامتازون نیز)
GSK-3؟ (بازخورد نظارتی)
فعال سازی HDAC
Halofuginone
هوموسیستئین (ALCAR می تواند این هوموسیستئین را منفی کند و سطح پایین NRF2 را منجر شود)
IL-24
Keap1
MDA-7
NF؟ B
اکراتوکسین A (گونه های آسپرژیلوس و پنسیلیوم)
پروتئین لوکمی Promyelocytic
p38
p53
p97
Retinoic acid receptor alpha
سلنیت
SYVN1 (Hrd1)
مهار STAT3 (مانند کریپوتانسینون)
تستوسترون (و تستوسترون پروپیونات، اگرچه TP در داخل بینی ممکن است NRF2 را افزایش دهد)
Trecator (Ethionamide)
Trx1 (از طریق کاهش Cys151 در Keap1 یا Cys506 در ناحیه NLS از Nrf2)
ترولکس
Vorinostat
کمبود روی (باعث می شود بدتر در مغز)
NoF2 مکانیسم عمل
استرس اکسیداتیو از طریق CUL3 آغاز می شود که در آن NRF2 از KEAP1، مهار کننده منفی، بعدا وارد هسته این سلول ها می شود، تحریک رونویسی ARE ها، تبدیل سولفید ها به دی سولفید ها و تبدیل آنها به ژن های آنتی اکسیدان بیشتر، منجر به تنظیم مقادیر آنتی اکسیدان ها می شود. به عنوان GSH، GPX، GST، SOD، و غیره. بقیه این را می توان در لیست زیر مشاهده کرد:
افزایش AKR
افزایش می یابد
ATF4 را افزایش می دهد
Bcl-xL را افزایش می دهد
Bcl-2 را افزایش می دهد
افزایش BDNF
BRCA1 را افزایش می دهد
c-Jun افزایش می یابد
CAT افزایش می یابد
cGMP را افزایش می دهد
CKIP-1 را افزایش می دهد
CYP450 را افزایش می دهد
Cul3 را افزایش می دهد
GCL افزایش می یابد
GCLC را افزایش می دهد
GCLM را افزایش می دهد
GCS را افزایش می دهد
افزایش GPx
GR افزایش می یابد
GSH افزایش می یابد
افزایش GST
HIF1 را افزایش می دهد
HO-1 را افزایش می دهد
HQO1 افزایش می یابد
HSP70 افزایش می یابد
IL-4 را افزایش می دهد
IL-5 را افزایش می دهد
IL-10 را افزایش می دهد
IL-13 را افزایش می دهد
K6 را افزایش می دهد
K16 را افزایش می دهد
K17 را افزایش می دهد
افزایش MHH
Mrp2-5 را افزایش می دهد
NADPH را افزایش می دهد
افزایش نمره 1
NQO1 را افزایش می دهد
افزایش PPAR-آلفا
Prx افزایش می یابد
p62 را افزایش می دهد
Sesn2 را افزایش می دهد
Slco1b2 را افزایش می دهد
sMafs را افزایش می دهد
SOD افزایش می یابد
Trx افزایش می یابد
افزایش Txn (d)
UGT1 (A1 / 6) را افزایش می دهد
افزایش VEGF
کاهش ADAMTS (4 / 5)
کاهش آلفا SMA
کاهش ALT
AP1 را کاهش می دهد
کاهش AST
کاهش Bach1
COX-2 را کاهش می دهد
کاهش DNMT
کاهش FASN
کاهش FGF
کاهش HDAC
IFN را کاهش می دهد-؟
کاهش IgE
کاهش IGF-1
IL-1b کاهش می یابد
IL-2 را کاهش می دهد
IL-6 را کاهش می دهد
IL-8 را کاهش می دهد
IL-25 را کاهش می دهد
IL-33 را کاهش می دهد
کاهش iNOS
کاهش LT
کاهش Keap1
MCP-1 را کاهش می دهد
MIP-2 را کاهش می دهد
کاهش MMP-1
کاهش MMP-2
کاهش MMP-3
کاهش MMP-9
کاهش MMP-13
کاهش NfkB
NO را کاهش می دهد
کاهش SIRT1
کاهش TGF-b1
کاهش TNF-alpha
کاهش Tyr
VCAM-1 را کاهش می دهد
کدگذاری شده از ژن NFE2L2، NRF2 یا عامل 2 وابسته به erythroid 2 هسته ای عامل فاکتور رونویسی در زیپ پایه لوسین یا bZIP است که از خانواده Cap'n'Collar یا ساختار CNC استفاده می کند.
این ترویج آنزیم های نیتریک، آنزیم های بیوتانسفرانس و انتقال دهنده های Xenobiotic efflux است.
این یک تنظیم کننده ضروری در القا anti آنتی اکسیدان فاز II و ژن های آنزیم سم زدایی است که سلول ها را از آسیب ناشی از استرس اکسیداتیو و حملات الکتروفیل محافظت می کند.
در طی شرایط هوموستاتیک، Nrf2 در سیتوزول از طریق اتصال بدن به دامنه N-terminal از Nrf2 یا پروتئین وابسته به ECH-کلش یا Keap1، نیز نامیده می شود که INrf2 یا مهارکننده Nrf2 نیز مهار فعال سازی Nrf2 نامیده می شود، جدا شده است.
همچنین ممکن است توسط سلنئوپروتئین تیرودوکسین ردوکتاز 1 یا TrxR1 پستانداران کنترل شود که به عنوان یک تنظیم کننده منفی عمل می کند.
پس از آسیب پذیری به استرسورهای الکتروفیلی، Nrf2 از Keap1 جدا می شود، انتقال به هسته، جایی که بعد از آن با تعدادی از پروتئین های تنظیم کننده رونویسی تقسیم می شود.
تعاملات مکرر شامل آنهایی هستند که مسئولین رونویسی Jun و Fos هستند که می توانند اعضای خانواده پروتئین فعال کننده پروتئین رونویسی باشند.
پس از dimerization، این مجتمع ها سپس به اجزای پاسخ دهنده آنتی اکسیدان / الکتروفیل ARE / EpRE متصل می شوند و رونویسی را فعال می کنند، همانطور که در مجامع Jun-Nrf2 درست است یا رونویسی را خنثی می کنند، بسیار شبیه به مجموعه Fos-Nrf2.
موقعیت ARE که مهار یا مهار می شود، تعیین می کند که کدام ژن ها توسط این متغیرها کنترل می شوند.
هنگامی که ARE فعال می شود:
فعال سازی سنتز آنتی اکسیدان ها قادر به سم زدایی از ROS مانند کاتالاز ، سوپراکسید-دیسموتاز یا SOD ، GSH-پراکسیدازها ، GSH- ردوکتاز ، GSH-ترانسفراز ، NADPH-کینون اکسیدوراوکتاز یا NQO1 ، سیستم سیتوکروم P450 مونوکسی ردوکسین ، ثوکسی اکسیدوژنازین ، تیوکسی اکسیدوژنازین ، تیوکسی اکسیدوژنازین ، تیوکسی اکسیدوژنازین ، سیستم ردوکتاز و HSP70.
فعال سازی این سنتاز GSH اجازه رشد قابل توجه درجه داخل سلولی GSH را می دهد که کاملاً محافظ است.
افزایش این سنتز و درجه آنزیم فاز II مانند UDP-گلوکورونوزیلت ترانسفراز، N-استیل ترانسفراز و سولفوترانسفراز.
تنظیم مقادیر HO-1، که یک گیرنده واقعا محافظ با رشد بالقوه CO است که در ارتباط با NO اجازه می دهد تا واجدولت سلول های ایسکمیک باشد.
کاهش بیش از حد آهن از طریق افزایش فریتین و بیلی روبین به عنوان یک آنتی اکسیدان لیپوفیلی. هر دو پروتئین فاز II همراه با آنتی اکسیدان ها قادر به رفع استرس اکسیداتیو مزمن و همچنین احیا یک سیستم بازسازی منظم طبیعی می باشند.
GSK3؟ تحت مدیریت AKT و PI3K ، فسفریلات Fyn منجر به محلی سازی هسته ای Fyn می شود ، که Fyn فسفریله Nrf2Y568 منجر به صادرات هسته ای و تخریب Nrf2 می شود.
NRF2 همچنین پاسخ TH1 / TH17 را کاهش می دهد و پاسخ TH2 را غنی می کند.
مهار کننده های HDAC منجر به مسیر سیگنالینگ nrf2 شده و تنظیم شده است که NOF2 در پایین دست HO-1، NQO1 و زیر واحد کاتالیزوری لیگاز گلوتامات و لیستاز گلوتامات یا GCLC، با محدود کردن Keap1 و تشویق جداسازی Keap1 از Nrf2، انتقال Nf2 هسته و Nrf2 -برچسب بودن
Nrf2 شامل نیمه عمر حدود 20 دقیقه در شرایط پایه است.
در حال کاهش IKK هستید؟ استخر از طریق اتصال Keap1 باعث کاهش I؟ B؟ تخریب و ممکن است مکانیسم گریزی باشد که ثابت می شود فعال شدن Nrf2 باعث مهار فعال سازی NF؟ B می شود.
Keap1 همیشه نباید تنظیم شود تا NRF2 بتواند عمل کند مانند chlorophyllin، blueberry، ellagic acid، astaxanthin و polyphenols چای ممکن است NRF2 و KEAP1 را در 400 درصد افزایش دهد.
Nrf2 از طریق مدت stearoyl CoA desaturase، یا SCD، و سیتریت لیاز یا CL، منفی را تنظیم می کند.
ژنتیک
KEAP1
rs1048290
آلل C نشان دهنده خطر قابل ملاحظه و اثر محافظتی در برابر صرع مقاوم در برابر دارو (DRE)
rs11085735 (من AC هستم)
در ارتباط با میزان کاهش عملکرد ریه در LHS
MAPT
rs242561
آلل T - آلل محافظتی برای اختلالات پارکینسونی - پیوند NRF2 / sMAF قوی تر داشت و با MAPT mRNA بالاتر در مناطق مختلف مغز، از جمله قشر مخچه (CRBL)، قشر موقت (TCTX)، ماده سفید سفید اینترلوووبار (WHMT)
NFE2L2 (NRF2)
rs10183914 (من CT هستم)
آلل T - افزایش سطح پروتئین Nrf2 و تاخیر در سن شروع پارکینسون به مدت چهار سال
rs16865105 (من AC هستم)
آلل C - دارای خطر بالایی از بیماری پارکینسون بود
rs1806649 (من CT هستم)
آلل C - شناخته شده است و ممکن است مربوط به علت سرطان پستان باشد.
همراه با افزایش خطر پذیرش بیمارستان در طول دوره های PM10 بالا
rs1962142 (من GG هستم)
آلل T - با سطح پایین بیان بیان NRF2 سیتوپلاسمی (P = 0.036) و بیان منفی sulfiredoxin (P = 0.042)
آلل - محافظت از جریان خون ساعد (FEV) کاهش می یابد (میزان انبساط مجاز در یک ثانیه) در رابطه با وضعیت سیگار کشیدن (p = 0.004)
rs2001350 (من TT)
T آلل - محافظت از کاهش FEV (حجم انبساط مجاز در یک ثانیه) در رابطه با سیگار کشیدن سیگار (P = 0.004)
rs2364722 (من AA هستم)
آلل - محافظت از کاهش FEV (حجم انبساط مجاز در یک ثانیه) در رابطه با وضعیت سیگار کشیدن (p = 0.004)
rs2364723
آلل C - به طور قابل توجهی کاهش FEV در سیگاری های ژاپنی با سرطان ریه
rs2706110
آلل G - نشان دهنده خطر قابل توجهی و اثر محافظتی در برابر صرع مقاوم در برابر دارو (DRE)
آلل های AA - بیان شد KEAP1 به طور قابل توجهی کاهش یافته است
آلل های AA - با افزایش خطر ابتلا به سرطان سینه همراه بود (P = 0.011)
rs2886161 (من TT)
آلل T - همراه با بیماری پارکینسون
rs2886162
یک آلل - با بیان NRF2 پایین همراه بود (P = 0.011؛ OR ، 1.988؛ CI ، 1.162 3.400) و ژنوتیپ AA با بقای بدتری همراه بود (P = 0.032؛ HR ، 1.687؛ CI ، 1.047)
rs35652124 (من TT)
آلل - همراه با افزایش سن با شروع بیماری آلزایمر بیماری آلزایمر در برابر پارکینسون
آلل C - پروتئین NRF2 را افزایش داده است
آلل T - دارای پروتئین NRF2 کمتر و خطر بیشتر بیماری قلبی و فشار خون بود
rs6706649 (من CC هستم)
آلل C - پروتئین NRF2 پایین تر داشته و خطر بیماری پارکینسون را افزایش می دهد
rs6721961 (من GG هستم)
آلل T - دارای پروتئین NRF2 پایین تر بود
آلل TT - ارتباط بین مصرف سیگار در سیگاری های سنگین و کاهش کیفیت مایع منی
آلل TT - با افزایش خطر سرطان پستان همراه بود [0.008/4.656 = P؛ OR ، 1.350 ؛ فاصله اطمینان (CI) ، 16.063-2] و آلل T با میزان کمی بیان پروتئین NRF0.0003 (2.420 = P ، OR ، 1.491 ، CI ، 3.926-1) و بیان SRXN0.047 منفی (P = 1.867؛ OR) همراه بود. 1.002 ؛ CI = 3.478 XNUMX)
آلل T - آلل نیز به طور اسمی مرتبط با مرگ و میر 28 روز مرتبط با ALI پس از سندرم پاسخ التهابی سیستمیک
T آلل - محافظت از کاهش FEV (حجم انبساط مجاز در یک ثانیه) در رابطه با سیگار کشیدن سیگار (P = 0.004)
آلل G - همراه با افزایش خطر ابتلا به ALI پس از آسیب عمده در اروپا و آفریقایی-آمریکایی (نسبت شانس، OR 6.44، 95٪ اطمینان فواصل
آلل های AA - همراه با آسم ناشی از عفونت
آلل های AA - نشان دهنده ژن NRF2 به طور قابل توجهی کاهش یافته و در نتیجه افزایش خطر ابتلا به سرطان ریه، به ویژه افرادی که تا به حال سیگار کشیدن
آلل های AA - به طور معنی داری بیشتر برای توسعه T2DM (OR 1.77، 95٪ CI 1.26، 2.49، P = 0.011) نسبت به کسانی که با ژنوتیپ CC
آلل های AA - ارتباط قوی بین بهبود زخم و سمیت های تابش اشعه (با ریسک قابل ملاحظه ای برای ایجاد اثرات ناگهانی در آفریقایی-آمریکایی ها با روند در قفقاز)
همراه با استروژن خوراکی و خطر ترومبوآمبولی وریدی در زنان یائسه
rs6726395 (من AG هستم)
یک آلل - از کاهش FEV1 (حجم انبساط اجباری در یک ثانیه) در رابطه با سیگار کشیدن (p = 0.004) محافظت شده
آلل - همراه با FEV1 به طور قابل توجهی کاهش یافته است در سیگاری های ژاپنی با سرطان ریه
آلل GG - سطح NRF2 بالاتری داشت و خطر ابتلا به دژنراسیون ماکولا را کاهش داد
آلل GG - دارای بقای بیشتر با Cholangiocarcinoma بود
rs7557529 (من CT هستم)
آلل C - همراه با بیماری پارکینسون
استرس اکسیداتیو و سایر عوامل استرس زا ممکن است موجب آسیب سلولی شود که در نهایت منجر به مسائل مختلف سلامتی شود. مطالعات انجام شده نشان داده است که فعال سازی Nrf2 می تواند مکانیزم آنتی اکسیدانی محافظ بدن انسان را ارتقاء دهد، اما محققان بر این عقیده اند که بیش از حد بیان Nrf2 می تواند خطرات فوق العاده ای نسبت به سلامت و سلامت کلی داشته باشد. انواع مختلف سرطان همچنین می توانند با بیش فعال سازی Nrf2 رخ دهند.
دکتر الکس جیمنز DC، CCST Insight
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
طبق مطالعات تحقیقاتی ، Nrf2 ، یک فاکتور اساسی رونویسی است که مکانیسم های آنتی اکسیدانی محافظ سلول ها را برای سم زدایی از بدن انسان فعال می کند. بیان بیش از حد Nrf2 ، می تواند باعث مشکلات سلامتی شود. دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
دکتر الکس جیمنز سرپرستی می کند
بحث در مورد مبحث اضافی: درد کمر حاد
درد پشتاین یکی از مهمترین دلایل معلولیت و روزهای از دست رفته در کار در سراسر جهان است. کمردرد دومین دلیل شایع مراجعه به مطب توسط پزشکان است که بیشتر از عفونت های دستگاه تنفسی فوقانی است. تقریباً 80 درصد مردم حداقل یک بار در طول زندگی خود درد کمر را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده است که از استخوان ها ، مفاصل ، رباط ها و ماهیچه ها در میان سایر بافت های نرم تشکیل شده است. آسیب ها و / یا شرایط وخیم ، ماننددیسک های فتق دیسک، در نهایت می تواند منجر به علائم درد پشت شود. آسیب های ورزشی یا آسیب های ناشی از تصادفات خودرو اغلب اغلب علت درد پشت هستند، اما گاهی اوقات ساده ترین حرکات می توانند نتایج دردناکی داشته باشند. خوشبختانه، گزینه های درمان جایگزین، مانند مراقبت از کیهان پراکسی، می تواند به کاهش درد در استفاده از تنظیمات ستون فقرات و دستکاری دست کمک کند و در نهایت بهبود تسکین درد را کاهش دهد.
بسیاری از مطالعات تحقیقاتی موجود در زمینه سرطان، متخصصین بهداشتی را درک کرده اند که چگونه بدن را از بین می برند. محققان با تجزیه و تحلیل ژن های کنترل شده در سلول های تومور، کشف کردند 2 عامل مربوط به 2 هسته ای سیگنالینگ، بهتر است به عنوان Nrf2 شناخته شود. NRF2 عامل مهم رونویسی است که بدن انسان را فعال می کند مکانیسم های آنتی اکسیدانی محافظتی برای تنظیم اکسیداسیون از عوامل خارجی و داخلی برای جلوگیری از افزایش سطوح استرس اکسیداتیو.
اصول Nrf2
NRF2 برای حفظ سلامت و سلامتی اساسی ضروری است زیرا هدف اصلی آن تنظیم این است که چگونه همه چیزهایی را که روزانه در معرض آن هستیم مدیریت کنیم و بیمار نشویم. فعال سازی NRF2 در سیستم سم زدایی فاز II نقش دارد. سم زدایی فاز II رادیکال های آزاد لیپوفیلی ، محلول در چربی را گرفته و آنها را به مواد دفع کننده آب دوست ، یا محلول در آب تبدیل می کند در حالی که متابولیت ها و مواد شیمیایی فوق العاده واکنش دهنده را غیرفعال می کند. مرحله اول
فعال شدن NRF2 باعث کاهش اکسیداسیون و التهاب بدن انسان از طریق اثر مهاری می شود. برای تحریک NRF2، واکنش التهابی به دلیل اکسیداسیون باید انجام شود تا سلول ها یک واکنش تطبیقی تولید کنند و آنتی اکسیدان ها مانند گلوتاتیون ایجاد کنند. اساسا برای شکستن اصل Nrf2، استرس اکسیداتیو NRF2 را فعال می کند که پس از آن یک پاسخ آنتی اکسیدانی را در بدن انسان فعال می کند. عملکرد NRF2 برای تعادل سیگنال redox یا تعادل سطح اکسیدان و آنتی اکسیدان در سلول است.
تصویری عالی از چگونگی عملکرد این فرآیند با ورزش نشان داده می شود. از طریق هر تمرین ، عضله سازگار می شود تا بتواند یک جلسه تمرین دیگر را در خود جای دهد. اگر NRF2 به دلیل عفونت های مزمن یا افزایش قرار گرفتن در معرض سموم ، کمتر یا بیش از حد بیان شود ، که ممکن است در بیمارانی که سندرم پاسخ التهابی مزمن یا CIRS دارند مشاهده شود ، ممکن است مسائل بهداشتی به دنبال فعال شدن NRF2 بدتر شود. بیش از همه ، اگر DJ-1 بیش از حد اکسید شود ، فعال سازی NRF2 به سرعت خاتمه می یابد.
اثرات فعال سازی NRF2
فعال شدن NRF2 به شدت در ریه ها، کبد و کلیه بیان می شود. فاکتور 2 یا NRF2 مرتبط با اریتروپوئن هستهای 2 اغلب با جلوگیری از افزایش میزان اکسیداسیون در بدن انسان که می تواند به استرس اکسیداتیو منجر شود عمل می کند. Activation Nrf2 می تواند به درمان بسیاری از مسائل مربوط به سلامت کمک کند، با این حال، بیش از حد activation از Nrf2 ممکن است مشکلات مختلف را تحت الشعاع، که در زیر نشان داده شده است.
فعال سازی دوره ای Nrf2 می تواند کمک کند:
پیری (یعنی طول عمر)
Autoimmunity و التهاب کلی (یعنی آرتریت، اوتیسم)
سرطان و حفاظت شیمیایی (به عنوان مثال نوردهی EMF)
افسردگی و اضطراب (یعنی PTSD)
قرار گرفتن در معرض مواد مخدر (الکل، NSAIDs)
ورزش و استقامت عملکرد
بیماری های قلب (یعنی SIBO، دیس Biosis، کولیت اولسراتیو)
بیماری کلیه (یعنی آسیب حاد کیست، بیماری مزمن کلیوی، نفریت لوپوس)
بیماری کبد (یعنی بیماری کبدی الکلی، هپاتیت حاد، بیماری کبد چرب غیر آلرژی، استئاتو هپاتیت غیرالکلی، سیروز)
بیماری ریه (یعنی آسم، فیبر)
بیماری های متابولیک و عروقی (یعنی آترواسکلروز، فشار خون بالا، سکته مغزی، دیابت)
عصب دهی (یعنی آلزایمر، پارکینسون، هانتینگتون و ALS)
پیوند قلب (در حالی که NRF2 باز ممکن است بد باشد، NRF2 می تواند در تعمیر کمک کند)
هپاتیت C
نفریت (موارد شدید)
ویتیلیگو
علاوه بر این ، NRF2 می تواند به ایجاد مکمل های غذایی خاص ، داروها ، و داروها کمک کند. بسیاری از مکمل های طبیعی نیز می توانند باعث تحریک NRF2 شوند. از طریق مطالعات تحقیقاتی فعلی ، محققان نشان داده اند که تعداد زیادی از ترکیباتی که زمانی اعتقاد بر این بود که آنتی اکسیدان هستند ، واقعاً طرفدار اکسیدان هستند. این به این دلیل است که تقریباً همه آنها برای عملکرد به NRF2 نیاز دارند ، حتی مکمل هایی مانند کورکومین و روغن ماهی. به عنوان مثال ، نشان داده شد که کاکائو در موشهایی که ژن NRF2 دارند ، اثرات آنتی اکسیدانی ایجاد می کند.
راه هایی برای فعال سازی NRF2
در مورد بیماری های عصبی مانند بیماری آلزایمر، بیماری پارکینسون، سکته مغزی و یا حتی بیماری های خود ایمنی، احتمالا بهتر است که Nrf2 تنظیم شود، اما به آرامی. مخلوط کردن فعال کننده NRF2 همچنین ممکن است اثر افزایشی یا هم افزایی داشته باشد، زیرا گاهی اوقات می توان وابسته به دوز باشد. راه های بالا برای افزایش بیان Nrf2 در زیر ذکر شده است:
HIST (ورزش) + CoQ10 + Sun (این همکاری بسیار خوب است)
Broccoli Sprouts + LLLT در سر و روده من
Butyrate + Super Coffee + صبح خورشید
طب سوزنی (این یک روش جایگزین، طب سوزنی لیزر نیز ممکن است استفاده شود)
خشک دهن
Cannabidiol (CBD)
مردانه شیر + ملاتونین
اسید آلفا لیپوئیک + DIM
کرم چوب
PPAR گاما فعال سازی
فهرست جامع زیر حاوی بیش از 350 راه های دیگر برای فعال سازی Nrf2 از طریق رژیم غذایی، شیوه زندگی و دستگاه ها، پروبیوتیک ها، مکمل ها، گیاهان و روغن ها، هورمون ها و انتقال دهنده های عصبی، داروها / داروها و مواد شیمیایی، عوامل مسیر / رونویسی، و همچنین راه های دیگر راهنمای مختصر در مورد چیزی که می تواند Nrf2 را ایجاد کند. به خاطر خلاصه ای از این مقاله، ما بیش از سایر مواد غذایی، مکمل های غذایی و ترکیبات 500 که می توانند Nrf2 را فعال کنند، خارج شده ایم. زیر در زیر ذکر شده است:
رژیم غذایی:
توت آکای
الکل (شراب قرمز بهتر است، به خصوص اگر چوب پنبه ای در آن وجود داشته باشد، زیرا آلکالوئید پرولوکتیکویک از پروتکاتچویک نیز می تواند NRF2 را فعال کند. به طور کلی، الکل توصیه نمی شود، گرچه مصرف حسی NRF2 را افزایش می دهد. مصرف مزمن ممکن است NRF2 را کاهش دهد.
جلبک (جلبک دریایی)
سیب ها
چای سیاه
آجیل برزیلی
پروتئین بروکلی (و دیگر ایزوتوسیانات ها، سولفورفان و همچنین سبزیجات cruciferous مانند بوک چوی که D3T دارند)
زغال اخته (0.6-10 گرم در روز)
هویج (فالکارینون)
فلفل کایین (کپسی سایین)
کرفس (Butylththalide)
چگا (بتولین)
چای بابونه
چیا
سیب زمینی چینی
Chokeberries (Aronia)
شکلات (تیره یا کاکائو)
دارچین
قهوه (مانند اسید کلرژنیک، Cafestol و Kahweol)
Cordyceps عصاره
ماهی (و سرخدار)
دانه کتان
سیر
گی (احتمالا)
زنجبیل (و کارتامونین)
Gojiberries
گریپ فروت (Naringenin - 50 mg / kg / d naringenin)
انگور
چای سبز
گیاهان و بتها ا
قلب پالم
هجیکی / واکام
شانه عسل
کیوی
سس ها
مرد شیر
ماهووا
مانگوس (مانگفرین)
آلوریتس
شیر (بز، گاو - از طریق تنظیم میکروبیوم)
توت
روغن زیتون (Pomace - هیدروکسی تیرروسول و اسید Oleanolic)
اسیدهای چرب امگا 6 (لیپوکسین A4)
پرتقال نارنجی (مورن)
قارچ صدف
پاپایا - نوعی میوه استوایی
بادام زمینی
نخود کبوتر
انار (Punicalagin، Ellagic Acid)
پروپولیس (Pinocembrin)
سیب زمینی شیرین بنفش
رمبوتان (گرانین)
پیاز
Reishi
رودیولا رزا (سالیدروزید)
سبوس برنج (cycloartenyl ferulate)
برنج برنج
چای روبیوس
رزماری
افسانه
گلرنگ
روغن کنجد
سویا (و ایزوفلاون ها، داذیزین، جنیستین)
کدو
توت فرنگی
تارتاری گندم سیاه
آویشن
گوجه فرنگیها
دانه تونکا
زردچوبه
Wasabi
هندوانه
شیوه زندگی و دستگاه ها:
طب سوزنی و الکترواسکولار (از طریق آبشار کلاژن در ECM)
نور آبی
بازی های مغز (افزایش NRF2 در هیپوکامپ)
محدودیت کالری
سرد (دوش، باران، حمام یخ، دنده، کریوتراپی)
EMFs (فرکانس پایین، مانند PEMF)
ورزش (به نظر می رسد ورزش حاد مانند HIST یا HIIT برای ایجاد NRF2 مفیدتر است ، در حالی که ورزش طولانی تر باعث NRF2 نمی شود ، اما باعث افزایش سطح گلوتاتیون می شود)
رژیم غذایی چربی بالا (رژیم غذایی)
گرمای زیاد (سونا)
استنشاق هیدروژن و آب هیدروژن
درمان با اکسیژن بیش فعالی
درمان مادون قرمز (مانند جوووو)
ویتامین C داخل وریدی
کتوژنیک رژیم غذایی
ازن
سیگار کشیدن (توصیه نمی شود - به شدت سیگار کشیدن NRF2 افزایش می یابد، در صورت مصرف سیگار، NRF2 به طور فزاینده ای کاهش می یابد. اگر بخواهید سیگار بکشید، ریحان مقدس می تواند در برابر کمبود رژیم NRF2 محافظت کند)
خورشید (UVB و مادون قرمز)
پروبیوتیک ها:
Bacillus subtilis (fmbJ)
Clostridium butyricum (MIYAIRI 588)
لاکتوباسیلوس brevis
Lactobacillus casei (SC4 و 114001)
لاکتوباسیلوس کولینوئید
لاکتوباسیلوس گاسری (OLL2809، L13-Ia و SBT2055)
Lactobacillus helveticus (NS8)
Lactobacillus paracasei (NTU 101)
Lactobacillus plantarum (C88، CAI6، FC225، SC4)
لاکتوباسیلوس رامنوسوس (GG)
مکمل ها، گیاهان و روغن:
استیل L-کارنیتین (ALCAR) و کارنیتین
آلیسین
اسید آلفا لیپوئیک
عمتوفلئون
Andrographis paniculata
آگماتین
آپیگنین
آرژنین
آتریشاک (Cyanropicrin)
عاشاواندا
عظم کعب
Bacopa
Beefsteak (Isogemaketone)
بربرین
بتا کاریوفیلن
بیدنس پیلوزا
روغن زیتون سیاه (Thymoquinone)
کندر
بوتین
بوتیرات
Cannabidiol (CBD)
کاروتنیوئیدها (مانند بتاکاروتن [هم افزایی با لیکوپن - 2 15 میلی گرم در روز لیکوپن] ، فوکوکسانتین ، زئاکسانتین ، آستاگزانتین و لوتئین)
چیتراک
کلرلا
کلروفیل
گل داودی zawadskii
سینومومای
Sundew مشترک
مس
قبطی
CoQ10
کورکومین
دامینه
دن شنی / سرخ مریم (Miltirone)
اذعان
دایوسین
دونگ لیانگ کائو
دونگ کوای (جین سنگ زن)
Ecklonia Cava
EGCG
Elecampane / Inula
پوست Eucommia
اسید فرولیک
فیزتین
روغن ماهی (DHA / EPA - 3 1 گرم در روز روغن ماهی حاوی 1098 میلی گرم EPA و 549 میلی گرم DHA)
گالنگال
Gastrodin (Tian Ma)
گندیانا
گل شمعدانی
گینگو بلیبا (گینگزید B)
Glasswort
Gotu کلا
عصاره دانه انگور
مزرعه مودار
هریاتاکی (Triphala)
زالزالک
Helichrysum
هانا (Juglone)
هیبیسوس
هیگینامین
ریحان مقدس / Tulsi (اسید اورسولیک)
رازک
علف های هرز شاد (Icariin / Icariside)
Indigo Naturalis
آهن (توصیه نمی شود مگر اینکه ضروری باشد)
I3C
اشک کار
مورینگا اولیفرا (مانند کیمفرلف)
Inchinkoto (دسته کوچک موسیقی جاز Zhi Zi و Wormwood)
ریشه کودزو
ریشه شیرین بیان
لیندرا ریشه
لوتئولین (دوزهای بالا برای فعال شدن، دوزهای پایین تر NRF2 را در سرطان مهار می کند)
ماگنولیا
مانجیستا
Maximowiczianum (Acerogenin A)
آرنیکا مکزیکی
خار شیر
MitoQ
مو شیانگ
مکونا پرورینز
نیکوتامینید و NAD +
Panax جینسنگ
طعم خوشبو (مانند کریزین، اما کیریزین همچنین ممکن است NRF2 را از طریق تنظیم غلظت سیگنال PI3K / Akt کاهش دهد)
پاو دارکو (لاپاچو)
فلوترین
Piceatannol
PQQ
پروسیانیدین
تروستیلبن
پئوریا
Quercetin (فقط دوزهای بالا، دوزهای پایین تر NRF2 را مهار می کند)
کیان هو
شبدرچمنی
رزوراترول (Piceid و سایر فیتواستروژنها اساسا Knotweed)
باسن رز
چوب بلسان بنفش
روتین
کفپوش
ساراساپاریلا
Saururus chinensis
SC-E1 (گچ، یاس، شیرین بیان، Kudzu و Balloon Flower)
Schisandra
خود شفا (prunella)
Skullcap (Baicalin و Wogonin)
کله گوسفند
سی وو تانگ
سیدریتس
اسپیکنارد (آرلیا)
اسپیرولینا
مخمر سنت جان
سولفورفان
ساترلندیا
تائو هنگ سی وو
ثوری
تندر خدا وین (Triptolide)
توکوفرول ها (مانند ویتامین E یا لینالول)
Tribulus R
تو سیززی
TUDCA
ویتامین A (اگر چه رتینوئیدهای دیگر NRF2 را مهار می کنند)
ویتامین C (فقط با دوز بالا ، دوز کم باعث مهار NRF2 می شود)
Vitex / Chaste Tree
پونه سفید (Paeoniflorin از Paeonia lactiflora)
کرمwood (Hispidulin و Artemisinin)
شیائو یو وان (سرگردان آزاد و آسان)
یربا سانتا (Eriodictyol)
یوان زی (Tenuigenin)
Zi Cao (NRF2 را در سرطان کاهش می دهد)
روی
Ziziphus Jujube
هورمون ها و انتقال دهنده های عصبی:
آديپونکتين
آدروپین
استروژن (اما ممکن است NRF2 را در بافت پستان کاهش دهد)
ملاتونین
پروژسترون
اسید کینولینیک (در واکنش محافظتی برای جلوگیری از اکسیدوتوکسید)
سروتونین
هورمون های تیروئید مانند T3 (می توانند NRF2 را در سلول های سالم افزایش دهند، اما آن را در سرطان کاهش می دهند)
ویتامین D
مواد مخدر / داروها و مواد شیمیایی:
استامینوفن
استازولامید
آملودیپین
آرونوفین
باردكسولون متیل (BARD)
بنزنیدازول
BHA
CDDO-imidazolide
سفتریاکسون (و آنتی بیوتیک بتالاکتام)
Cialis
دگزامتازون
دیپران (پروپوفول)
Eriodictyol
Exendin-4
ایزتییب
فلورید
فومارات
HNE (اکسید شده)
Idazoxan
آرسنیک معدنی و آرسنیت سدیم
JQ1 (ممکن است NRF2 را نیز مهار کند، ناشناخته است)
Letairis
ملپالان
متازولامید
متیلن آبی
نیفدیپین
NSAIDs
اولتیپراس
PPI ها (مانند امپرازول و لانسپراازول)
Protandim - نتایج عالی در in vivo، اما ضعیف / غیر فعال در فعال شدن NRF2 در انسان
پروبوکول
راپامایسین
Reserpine
روتنیم
سیاتکسنتان
استاتین ها (مانند لیپیتور و سیمواستاتین)
تاموکسی فن
تانگ لو نینگ
tBHQ
Tecfidera (دی متیل فومارات)
THC (نه به عنوان قوی CBD)
تئوفیلین
Umbelliferone
اسید اورستوکسو هیکول (UDCA)
وراپامیل
ویاگرا
4-Acetoxyphenol
مسیرهای مسیر / عوامل رونویسی:
فعال سازی 7 nAChR
AMPK
بیلیروبین
CDK20
CKIP-1
CYP2E1
EAATs
گانکیرین
گرمیلی
GJA1
H-ferritin ferroxidase
مهار کننده های HDAC (مانند اسید والپروئیک و TSA، اما می توانند ناپایداری NRF2 را ایجاد کنند)
پروتئین شوک گرما
IL-17
IL-22
کلوتو
اجازه دهید 7 (رها کردن mBach1 RNA)
MAPK
پذیرش مایکل (اکثر)
میر 141
میر 153
miR-155 (همچنین رهاسازی mBach1 RNA را قطع می کند)
miR-7 (در مغز، به کمک سرطان و اسکیزوفرنیا کمک می کند)
Notch1
استرس اکسیداتیو (مانند ROS، RNS، H2O2) و Electrophiles
PGC-1؟
PKC دلتا
PPAR-گاما (اثرات سینرژیک)
مهار گیرنده سیگما-1
SIRT1 (NRF2 را در مغز و ریه افزایش می دهد، اما ممکن است به طور کلی آن را کاهش دهد)
SIRT2
SIRT6 (در کبد و مغز)
SRXN1
مهار TrxR1 (از بین بردن یا تخریب)
پروپوپورفیرین روی
4-HHE
دیگر:
آنکافلاوین
پنبه نسوز
آوایینس ها
Bacillus amyloliquefaciens (مورد استفاده در کشاورزی)
مونواکسید کربن
دافنیتین
کاهش گلوتاتیون (احتمالاً کاهش 80٪ -90٪)
تناسب اندام
هپاتیت C
هرپس (HSV)
خاکستر هندی
ریشه Indigowoad
Isosalipurposide
Isorhamentin
موناسن
Omaveloxolone (قوی، همانند RTA-408)
PDTC
کمبود سلنیوم (کمبود سلنیوم می تواند NRF2 را افزایش دهد)
نشاسته سیبری
Sophoraflavanone G
Tadehagi triquetrum
Toona sinensis (7-DGD)
ترومپت گل
63171 و 63179 (قوی)
عامل سیگنال 2 وابسته به اریتروتین 2، که به بهترین وجه نام اختصاری Nrf2 شناخته می شود، فاکتور رونویسی است که نقش مهمی در تنظیم سازوکارهای آنتی اکسیدانی محافظ بدن انسان، به ویژه برای کنترل استرس اکسیداتیو دارد. در حالی که افزایش سطوح استرس اکسیداتیو میتواند Nrf2 را فعال کند، اثرات آن از طریق وجود ترکیبات خاص به شدت افزایش مییابد. بعضی از غذاها و مکمل ها به بهبود عملکرد دستگاه Nof2 در بدن انسان کمک می کنند ایزوتیوسیانات سولفورفان از شاخه های بروکلی دکتر الکس جیمنز DC، CCST Insight
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
طبق بسیاری از مطالعات تحقیقاتی فعلی ، مسیر سیگنالینگ فاکتور 2 مربوط به اریتروئید هسته ای ، معروف به Nrf2 ، یک فاکتور اساسی رونویسی است که مکانیسم های آنتی اکسیدانی محافظ سلول را فعال می کند تا بدن انسان را از هر دو عامل خارجی و داخلی سم زدایی کند و از افزایش سطح استرس اکسیداتیو دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
دکتر الکس جیمنز سرپرستی می کند
بحث در مورد مبحث اضافی: درد کمر حاد
درد پشتاین یکی از مهمترین دلایل معلولیت و روزهای از دست رفته در کار در سراسر جهان است. کمردرد دومین دلیل شایع مراجعه به مطب توسط پزشکان است که بیشتر از عفونت های دستگاه تنفسی فوقانی است. تقریباً 80 درصد مردم حداقل یک بار در طول زندگی خود درد کمر را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده است که از استخوان ها ، مفاصل ، رباط ها و ماهیچه ها در میان سایر بافت های نرم تشکیل شده است. آسیب ها و / یا شرایط وخیم ، ماننددیسک های فتق دیسک، می تواند در نهایت به علائم کمردرد منجر شود. آسیب های ورزشی یا صدمات ناشی از تصادفات اتومبیل اغلب شایعترین علت کمر درد است ، با این حال ، گاهی اوقات ساده ترین حرکات می تواند نتایج دردناکی داشته باشد. خوشبختانه ، گزینه های درمانی جایگزین مانند مراقبت های کایروپراکتیک ، می توانند با استفاده از تنظیمات ستون فقرات و دستکاری های دستی ، درد کمر را کاهش دهند و در نهایت تسکین درد را بهبود بخشند.
استرس اکسیداتیو عامل مهمی در توسعه انواع مسائل مربوط به سلامت، از جمله سرطان، بیماری قلبی، دیابت، پیری سریع و تولید عصبی است. برای محافظت از بدن انسان از سطوح بالای استرس اکسیداتیو، می توان از غذاهای غنی از آنتی اکسیدان، گیاهان و مکمل ها استفاده کرد. مطالعات انجام شده اخیر نشان داده است که مسیر ژن Nrf2 می تواند اثرات آنتی اکسیدان ها را تقویت کند. این مزایای Nrf2 در زیر توضیح داده شده است.
بدن را از نظر سموم محافظت می کند
NRF2 یک ماده ذاتی است که می تواند سلول ها را از ترکیبات مضر، داخلی و خارجی محافظت کند. NRF2 ممکن است به تقویت واکنش بدن انسان به داروها/داروها و سموم کمک کند و تولید پروتئین هایی را که به حذف ترکیباتی از سلول کمک می کنند، که به عنوان پروتئین های مرتبط با مقاومت چند دارویی یا MRP شناخته می شوند، بهبود می بخشد. استنشاق دود سیگار به ریه ها اجازه سم زدایی می دهد.
علاوه بر این، برای ریه ها ضروری است که از خود در برابر آلرژن ها، بیماری های ویروسی، اندوتوکسین های باکتریایی، هایپراکسی و آلاینده های مختلف محیطی محافظت کنند. با این حال، محرک ثابت Nrf2 می تواند سطح ماده ای به نام گلوتاتیون را در سراسر بدن انسان کاهش دهد. NRF2 همچنین ممکن است از کبد در برابر سمیت محافظت کند و می تواند از کبد در برابر سمیت کبدی آرسنیک محافظت کند. علاوه بر این، NRF2 از کبد و مغز در برابر مصرف الکل محافظت می کند. به عنوان مثال، Nrf2 می تواند در برابر سمیت استامینوفن محافظت کند.
مبارزه با التهاب و استرس اکسیداتیو
فعال شدن NRF2 می تواند در مبارزه با التهاب با کاهش سیتوکین های التهابی، مانند کسانی که در پسوریازیس وجود دارد، کمک کند. NRF2 همچنین ممکن است التهاب همراه با انواع مختلف مسائل مربوط به سلامت مانند آرتروز و فیبروز کبد، کلیه و ریه را کاهش دهد. NRF2 همچنین می تواند با کاهش سیتوکین های Th1 / Th17 و افزایش سیتوکین های TH2 به کنترل آلرژی ها کمک کند. این می تواند برای بیماری مانند آسم سودمند باشد.
NRF2 علاوه بر این از آسیب سلولی ناشی از نور آبی و UVA/UVB موجود در نور خورشید محافظت می کند. کمبود Nrf2 می تواند آفتاب سوختگی را بسیار آسان تر کند. یکی از دلایل این امر این است که NRF2 قادر به تنظیم کلاژن در پاسخ به اشعه UV است. محصولات نهایی گلیکاسیون پیشرفته یا AGEs در ایجاد بسیاری از مشکلات سلامتی از جمله دیابت و بیماریهای عصبی کمک میکنند. NRF2 می تواند استرس اکسیداتیو AGE ها را در بدن کاهش دهد. NRF2 همچنین ممکن است بدن انسان را از سطوح بالاتر استرس ناشی از گرما محافظت کند.
تقویت میتوکندریا و عملکرد ورزشی
NRF2 یک تقویت کننده میتوکندری است. فعال سازی NRF2 علاوه بر افزایش استفاده از اکسیژن یا سیترات و چربی به افزایش انرژی ATP برای میتوکندری کمک می کند. بدون NRF2، میتوکندری فقط توانایی عملکرد با قند یا گلوکز را دارد تا چربی. NRF2 همچنین برای توسعه میتوکندری از طریق فرآیندی به نام بیوژنز ضروری است. فعال سازی NRF2 برای استفاده از مزایای ورزش حیاتی است.
به دلیل فعالیت Nrf2 ، ورزش عملکرد میتوکندری را افزایش می دهد ، جایی که این نتیجه ممکن است با CoQ10 ، کوردیسپس و محدودیت کالری تقویت شود. ورزش متوسط یا ورزش حاد از طریق فعال سازی NRF1 باعث پیدایش میتوکندری و سنتز بالای سوپراکسید دیسموتاز یا SOD و هم اکسیژناز -1 یا HO-2 می شود. آلفا-لیپوئیک اسید ، or ALA و دن شن می توانند بیوژنز میتوکندری با واسطه NRF2 را تقویت کنند. علاوه بر این ، در مواردی که حذف NRF2 ورزش را مضر می کند ، whereNRF2 می تواند تحمل ورزش را نیز بهبود بخشد.
محافظت در برابر هیپوکسی
NRF2 نیز کمک می کند تا بدن انسان را از از دست دادن / کاهش اکسیژن سلولی، یک مسئله بهداشتی به نام هیپوکسی محافظت کند. افراد مبتلا به CIRS سطح اکسيژن را کاهش داده اند، زيرا NRF2 مانع آن است، و در نتيجه سطح VEGF، HIF1 و HO-1 کاهش مي يابد. به طور معمول، افراد سالم با هیپوکسی، miR-101 که برای ایجاد سلول های بنیادی مورد نیاز است، بیش از حد بیان می شود و مقدار NRF2 / HO-1 و VEGF / eNOS را افزایش می دهد، بنابراین جلوگیری از آسیب مغزی، اما به نظر نمی رسد در CIRS
هیپوکسی، که دارای HIF1 کم است، در CIRS نیز می تواند منجر به یک مانع از خونریزی خون ناشی از عدم تعادل NRF2 شود. Salidroside، واقع در Rhodiola، بر روی فعال سازی NRF2 عمل می کند و با افزایش سطح VEGF و HIF1 در بدن انسان کمک می کند تا با هیپوکسی. NRF2 همچنین می تواند در برابر ایجاد لاکتات در قلب محافظت کند. فعال شدن NRF2 همچنین ممکن است بیمار مبتلا به ارتفاع حرکت ناشی از هیپوکسی یا AMS را متوقف کند.
کند شدن پیری را کاهش می دهد
ترکیبات متعددی که ممکن است در مقادیر زیاد کشنده باشند ممکن است به دلیل xenohormesis از طریق NRF2، PPAR-گاما و FOXO، طول عمر را در مقادیر نسبتاً کوچک افزایش دهند. مقدار بسیار کمی از سموم، توانایی سلول را برای تجهیز بهتر برای دفعه بعدی که با یک سم به چالش میکشد، افزایش میدهد، با این حال، این تاییدی برای مصرف مواد شیمیایی سمی نیست.
یک مثال خوب از این فرآیند با محدودیت کالری است. NRF2 می تواند طول عمر سلول ها را با افزایش سطح میتوکندری و آنتی اکسیدان ها و همچنین کاهش توانایی سلول ها برای مرگ بهبود بخشد. NRF2 با افزایش سن کاهش می یابد زیرا NRF2 از مرگ سلول های بنیادی جلوگیری می کند و به بازسازی آنها کمک می کند. NRF2 در بهبود زخم نقش دارد.
تقویت سیستم عروقی
فعال سازی NRF2 به درستی با تولید سولفورفان انجام می شود و ممکن است در برابر بیماری های قلبی مانند فشار خون بالا یا فشار خون بالا و سخت شدن شریان ها و یا آترواسکلروز محافظت کند. NRF2 می تواند فعالیت آرامش بخش استیل کولین یا ACh را در سیستم عروقی و کاهش استرس ناشی از کلسترول افزایش دهد. Activation Nrf2 ممکن است قلب را تقویت کند، با این حال، بیش از حد فعال Nrf2 می تواند احتمال بیماری قلبی عروقی را افزایش دهد.
استاتین ها ممکن است منجر به بیماری قلبی عروقی شوند. NRF2 همچنین نقش مهمی در تعادل آهن و کلسیم ایفا می کند که ممکن است بدن انسان از افزایش سطح آهن جلوگیری کند. به عنوان مثال، Sirtuin 2 یا SIRT2 می تواند هوموستاز آهن را در سلول ها با فعال شدن NRF2 تنظیم کند که به نظر می رسد برای سطح سالم آهن مورد نیاز است. NRF2 همچنین می تواند به بیماری سلول های سقط یا SCD کمک کند. اختلال NRF2 ممکن است دلیلی برای اندوتوکسمی باشد مانند دیس Biosis یا فشار خون ناشی از لکتین. Nrf2 همچنین ممکن است بدن انسان را در مقابل آسیب های ناشی از آمفتامین به سیستم عروقی محافظت کند.
مبارزه با نوروپاتی
NRF2 می تواند در مقابل التهاب مغز، که معمولا به عنوان عفونت های عصبی شناخته می شود، محافظت کند. علاوه بر این، NRF2 می تواند با مجموعه ای از سیستم عصبی مرکزی، یا CNS، اختلالات، از جمله:
بیماری آلزایمر (AD) - استرس بتا آمیلوئید را بر روی میتوکندری کاهش می دهد
اسکلروز جانبی جانبی آمیوتروفیک (ALS)
بیماری هانتینگتون (HD)
مولتیپل اسکلروزیس (MS)
عصب بازسازی
بیماری پارکینسون (PD) - محافظت از دوپامین
آسیب نخاعی (SCI)
سکته مغزی (ایسکمیک و هموراژیک) - کمک به هیپوکسی
آسیب مغزی پس از حادثه (PTSD)
NRF2 کاهش التهاب عصبی را در نوجوانان مبتلا به اختلالات طیف اوتیسم یا ASD نشان داده است. Idebenone برخلاف التهاب عصبی به درستی با فعال کننده های NRF2 جفت می شود. NRF2 همچنین ممکن است سد خونی مغز یا BBB را بهبود بخشد. به عنوان مثال، فعال سازی NRF2 با اسید کارنوزیک به دست آمده از رزماری و مریم گلی می تواند از BBB عبور کرده و باعث نوروژنز شود. همچنین ثابت شده است که NRF2 فاکتور نوروتروفیک مشتق از مغز یا BDNF را افزایش می دهد.
NRF2 همچنین ظرفیت برخی مکملهای غذایی را برای ایجاد فاکتور رشد عصبی یا NGF تعدیل میکند، زیرا میتواند با تعدیل گیرندههای N-Methyl-D-Aspartate یا NMDA به مه مغزی و مشکلات ناشی از گلوتامات کمک کند. همچنین ممکن است استرس اکسیداتیو ناشی از کینولینیک اسید، که به آن QUIN گفته می شود، کاهش دهد. فعال سازی NRF2 می تواند در برابر تشنج محافظت کند و دوزهای زیاد می تواند آستانه تشنج را کاهش دهد. در دوزهای منظم تحریک، NRF2 میتواند تواناییهای شناختی را پس از تشنج با کاهش گلوتامات خارج سلولی در مغز و با توانایی آن در جذب سیستئین از گلوتامات و گلوتاتیون افزایش دهد.
افسردگی را کاهش می دهد
در افسردگی، طبیعی است که التهاب در مغز، به خصوص از قشر پرفرانتال و هیپوکامپ، و نیز کاهش BDNF، توجه شود. در برخی از نسخه های افسردگی، NRF2 می تواند علائم افسردگی را کاهش دهد با کاهش التهاب داخل مغز و افزایش سطح BDNF. توانایی Agmatine برای کاهش افسردگی با افزایش نورآدرنالین، دوپامین، سروتونین و BDNF در هیپوکمپ بستگی به فعال شدن NRF2 دارد.
دارای خواص ضد سرطان است
NRF2 به همان اندازه یک سرکوب کننده تومور است؛ به عنوان یک پروموتر تومور، اگر به آن عمل نکنید. NRF2 می تواند در برابر سرطان ناشی از رادیکال های آزاد و استرس اکسیداتیو محافظت کند، با این حال، بیان بیش از حد NRF2 در سلول های سرطانی نیز می تواند یافت شود. فعال سازی شدید NRF2 می تواند با انواع سرطان ها کمک کند. به عنوان مثال، Protandim می تواند سرطان پوست را با فعال شدن NRF2 کاهش دهد.
درد را کاهش می دهد
بیماری جنگ خلیج فارس و یا GWI، یک بیماری قابل توجهی که جانبازان جنگ خلیج فارس را تحت تاثیر قرار می دهد، مجموعه ای از علائم مزمن ناشناخته است که ممکن است شامل خستگی، سردرد، درد مفاصل، بیماری های گوارشی، بی خوابی، سرگیجه، بیماری های تنفسی و مشکلات حافظه باشد. NRF2 می تواند علائم GWI را کاهش دهد با کاهش التهاب هیپوکامپ و کلی، علاوه بر کاهش درد. NRF2 می تواند علاوه بر درد از آسیب عصب بدن و بهبود آسیب عصبی از نوروپاتی دیابتی کمک کند.
دیابت را بهبود می بخشد
سطوح بالای گلوکز، که بیشتر به عنوان hyperglycemia شناخته می شوند، سبب آسیب اکسیداتیو به سلول ها به علت اختلال عملکرد میتوکندری می شود. فعال سازی NRF2 ممکن است بدن انسان را در برابر آسیب هیپرگلیسمی به سلول محافظت کند، در نتیجه جلوگیری از مرگ سلولی. فعال شدن NRF2 می تواند علاوه بر حفاظت، بازگرداندن و افزایش عملکرد سلول های پانکراس بتا، در حالی که کاهش مقاومت به انسولین.
محافظت از بینایی و شنوایی
NRF2 می تواند در مقابل آسیب دیدگی چشم از رتینوپاتی دیابتی محافظت کند. همچنین ممکن است از ایجاد آب مروارید جلوگیری کند و از فتوگرامترها محافظت کند بر خلاف مرگ ناشی از نور. NRF2 گوش و حلق و بینی را نیز از استرس و از دست دادن شنوایی محافظت می کند.
ممکن است چاقی کمک کند
NRF2 ممکن است با چاقی در ابتدا به دلیل توانایی آن برای تنظیم متغیرهایی که در تجمع چربی در بدن انسان عمل می کند، کمک کند. فعال شدن NRF2 با سولفورفان می تواند مهار سنتز اسید چرب، یا FAS، و پروتئین های جدا سازی یا UCP را افزایش دهد و منجر به کاهش تجمع چربی و چربی قهوه ای بیشتر به عنوان چربی می شود که شامل میتوکندری های بیشتری است.
محافظت از گوشت
NRF2 با محافظت از هوموستاز میکروبیوم روده کمک می کند تا از روده محافظت کند. به طور مثال، پروبیوتیک های Lactobacillus باعث می شود NRF2 برای محافظت از روده از استرس اکسیداتیو استفاده شود. NRF2 همچنین می تواند به جلوگیری از کولیت اولسراتیو یا UC کمک کند.
محافظت از سازمان های جنسی
NRF2 می تواند از بیضه ها محافظت کند و از تعداد اسپرم در افراد مبتلا به دیابت جلوگیری کند. همچنین می تواند به اختلال نعوظ یا ED کمک کند. برخی از مکمل های تقویت کننده میل جنسی مانند Mucuna، Tribulus و Ashwaganda ممکن است عملکرد جنسی را از طریق فعال سازی NRF2 افزایش دهند. عوامل دیگری که NRF2 را تقویت میکنند، مانند نور خورشید یا جوانههای بروکلی نیز میتوانند به بهبود میل جنسی کمک کنند.
تنظیمات استخوان ها و عضلات
استرس اکسیداتیو ممکن است سبب کاهش تراکم استخوان و کاهش قدرت استخوانی شود که در پوکی استخوان طبیعی است. فعال سازی NRF2 می تواند توانایی بهبود آنتی اکسیدان ها در استخوان ها و محافظت در برابر پیری استخوان داشته باشد. NRF2 همچنین می تواند باعث از بین رفتن عضلات و افزایش دیستروفی عضلانی دوچن یا DMD شود.
دارای خواص ضد ویروسی است
آخرین اما نه کم اهمیت، فعال سازی NRF2 در نهایت می تواند به دفاع از بدن انسان در برابر چندین ویروس کمک کند. در بیماران مبتلا به ویروس دنگی، علائم در افرادی که سطوح NRF2 بیشتری نسبت به افرادی که درجات NRF2 کمتری داشتند، شدید نبود. NRF2 همچنین می تواند به افرادی که ویروس نقص ایمنی انسانی-1 یا HIV دارند کمک کند. NRF2 می تواند در برابر استرس اکسیداتیو ناشی از ویروس آدنو مرتبط، یا AAV و هلیکوباکتر پیلوری محافظت کند. در نهایت، ریشه Lindera ممکن است ویروس هپاتیت C را با فعال سازی NRF2 سرکوب کند.
Nrf2 یا عامل NF-E2 مرتبط با 2، یک عامل رونویسی در انسان است که بیان یک مجموعه خاص از ژن های آنتی اکسیدان و سم زدایی را تنظیم می کند. این مسیر سیگنالینگ به علت استرس اکسیداتیو فعال شده است، زیرا آنزیم های متعدد آنتی اکسیدان و سم زدایی کبد فاز II را برای بازگرداندن هوموتازیس در بدن انسان افزایش می دهد. انسانها در سراسر حالت تهوع و استفراغ و یا تعادل عمل می کنند. هنگامی که بدن با استرس اکسیداتی مواجه می شود، Nrf2 برای تنظیم اکسیداسیون و کنترل استرس ایجاد می کند. Nrf2 برای جلوگیری از مشکلات بهداشتی مرتبط با استرس اکسیداتیو ضروری است. دکتر الکس جیمنز DC، CCST Insight
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
00: 46: 06 - ایزوتوسیانات به عنوان گیتروژن.
هنگامی که بدن انسان با عوامل مضر داخلی و خارجی مانند سموم روبرو می شود ، سلول ها باید به سرعت توانایی آنتی اکسیدانی خود را برای مقابله با استرس اکسیداتیو تحریک کنند. از آنجا که مشخص شده است افزایش میزان استرس اکسیداتیو باعث ایجاد انواع مسائل بهداشتی می شود ، استفاده از فعال سازی Nrf2 برای استفاده از مزایای آن مهم است. دامنه اطلاعات ما محدود به مسائل مربوط به بهداشت عمل جراحی و نخاع است. برای بحث در مورد موضوع ، لطفاً از دکتر جیمنز سوال کنید یا با ما تماس بگیرید915-850-0900.
دکتر الکس جیمنز سرپرستی می کند
بحث در مورد مبحث اضافی: درد کمر حاد
درد پشتاین یکی از مهمترین دلایل معلولیت و روزهای از دست رفته در کار در سراسر جهان است. کمردرد دومین دلیل شایع مراجعه به مطب توسط پزشکان است که بیشتر از عفونت های دستگاه تنفسی فوقانی است. تقریباً 80 درصد مردم حداقل یک بار در طول زندگی خود درد کمر را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده است که از استخوان ها ، مفاصل ، رباط ها و ماهیچه ها در میان سایر بافت های نرم تشکیل شده است. به همین دلیل ، صدمات و یا شرایط وخیم مانند دیسک های فتق دیسک، می تواند در نهایت به علائم کمردرد منجر شود. آسیب های ورزشی یا صدمات ناشی از تصادفات اتومبیل اغلب شایعترین علت کمر درد است ، با این حال ، گاهی اوقات ساده ترین حرکات می تواند نتایج دردناکی داشته باشد. خوشبختانه ، گزینه های درمانی جایگزین مانند مراقبت های کایروپراکتیک ، می توانند با استفاده از تنظیمات ستون فقرات و دستکاری های دستی ، درد کمر را کاهش دهند و در نهایت تسکین درد را بهبود بخشند.
سولفورفان یک فیتوشیمیایی است که درون گروه ایزوتوسیانات از ترکیبات ارگنسی سولفات موجود در سبزیجات cruciferous مانند بروکلی، کلم، گل کلم و گلدان بروکسل یافت می شود. همچنین می توان در کیک بوک، کلم، کلث، سبز خردل و کاکائو پیدا کرد. مطالعات انجام شده نشان داده است که سولفورفان می تواند به جلوگیری از انواع مختلف سرطان کمک کند فعال سازی تولید Nrf2، یا فاکتور هسته ای مربوط به اریتروئید 2 ، یک فاکتور رونویسی است که مکانیسم های آنتی اکسیدانی محافظ را تنظیم می کند که پاسخ سلول به اکسیدان ها را کنترل می کند. هدف مقاله زیر توصیف عملکرد سولفورافان است.
چکیده
سیستم آنتی اکسیدانی KEAP1-Nrf2-ARE یک وسیله اصلی است که سلول ها به تنش های اکسیداتیو و زانوبیوتیک پاسخ می دهند. سولفورفان (SFN)، یک ایزوتوسیانات الکتروفیل مشتق شده از سبزیجات cruciferous، مسیر KEAP1-Nrf2-ARE را فعال می کند و در درمان بیماری هایی که استرس اکسیداتیو مزمن نقش اصلی ایتیولوژیک ایفا می کند، تبدیل به یک مولکول شده است. ما در اینجا نشان می دهیم که میتوکندری سلول های اپیتلیال رنگدانه شبکیه (RPE-1) که تحت درمان با SFN کشت می شوند، تحت فشار زیاد قرار می گیرند که مستقل از هر دو مهارکننده Nrf2 و KEAP1 مهار کننده سیتوپلاسمی آن است. در طی آپوپتوز، با ایجاد مهار تولید ذرات در میتوکندری گزارش شده است که فیوژن میتوکندری به عنوان سیتوپروتئینی محسوب می شود و به این ترتیب، مستقل از سیتوپروتئین سفتی سلول های تحت درمان با SFN نشان می دهد که تحت تاثیر آپوپتوزیس-استوروسورپین قرار دارند. به طور مکانیکی، SFN جذب و یا حفظ فاکتور شکنندگی محلول Drp2 را به میتوکندریها و پراکسیوم ها کاهش می دهد اما بر میزان فراوانی Drp1 تاثیر نمی گذارد. این داده ها نشان می دهد که خواص سودمند SFN فراتر از فعال شدن سیستم KEAP1-Nrf1-ARE می باشد و بازجویی های بیشتری را اعطا می کند با توجه به استفاده فعلی از این عامل در چندین آزمایش بالینی.
سولفورفان یک مهار کننده مستقل Nrf2 از شکافت هسته ای است
سولفورفان (SFN) یک ترکیب ایزوتیوسیانات است که در رژیم غذایی بیشتر از سبزیجات cruciferous [56] یافت می شود. این در گیاهان به عنوان یک واکنش زبو بیوتیک به شکارچیان از طریق انتشار vesicular آنزیم هیدرولیز myrosinase از سلول های آسیب دیده تولید می شود؛ این آنزیم گلوکوزینولات را به ایزوتایوسیانت [42] تبدیل می کند. در طول دو دهه گذشته، SFN به طور گسترده ای برای خواص ضد سرطان، آنتی اکسیدان و ضد میکروبی گزارش شده است [57]. بیشتر این اثربخشی به ظرفیت SFN مربوط است تا مسیر سیگنالینگ KEAP1-Nrf2-antioxidant element (ARE) را تعدیل کند، اگر چه فعالیت های اضافی این ترکیب شناسایی شده است، از جمله مهار فعالیت هیستون داستیلاز و پیشرفت چرخه سلولی [ 29] Nrf2 عامل فاکتور رونویسی آنتی اکسیدان است و در شرایط هوموتازیست، پایداری آن از طریق عمل مجدد Cylin3KEAP1 ubiquitin ligase [20] سیتوپلاسمی سکته می کند. به طور خاص، Nrf2 به ليگاز Cullin3KEAP1 با اتصال به آداپتور Substrate دايرهيک KEAP1 استخدام مي شود و سپس با زنجيرهاي polyUb اصلاح مي شود که فاکتور رونويسي را براي تخليه مداخله پروتئازوم هدف قرار مي دهند. این توافق قانونی نیمه عمر "Nrf2" را در سلول های غیر قابل تحمل محدود می کند به ~ 15 دقیقه [30]، [33]، [46]، [55]. KEAP1، یک پروتئین غنی از پروتئین، به عنوان یک سنسور بازآفرینی عمل می کند و تغییرات اکسیداتیو سیتین های بحرانی، به ویژه C151، از KEAP1 دیس فیتی Nrf2-KEAP1 از CUL3 و از این طریق مانع از تجزیه Nif2 می شود [ 8]، [20]، [55]. به طور قابل ملاحظه ای، SFN، و احتمالا دیگر activators Nrf2، تقلید از استرس اکسیداتیو را با تغییر C151 از KEAP1 مانند [21]. تثبیت Nrf2 اجازه می دهد تا انتقال آن به هسته ای که آن را بیان یک باتری از ژن های آنتی اکسیدان فاز II و سم زدایی است. Nrf2 به عناصر پروتئینی واکنش آنتی اکسیدان (ARE) از ژن های هدف شناخته شده آن متصل می شود، از طریق heterodimerization با پروتئین های کوچک Maf [19]. این سیستم یک پاسخ پویا و حساس به آنتی اکسیدان های غیر مستقیم مانند SFN، رادیکال های آزاد تولید شده توسط میتوکندری [16] یا سایر منابع فیزیولوژیک استرس اکسیداتیو [41] را ارائه می دهد.
میتوکندریا اندامهای پویا و سلولی هستند که تعدادی از توابع سلولی را که از تولید ATP و بافر کلسیم داخل سلولی تا تنظیم مجدد و آپوپتوز [13]، [49] تنظیم می شود. میتوکندریا منبع اصلی گونه های اکسیژن واکنشی (ROS) را درون سلول نشان می دهد. بنابراین تنظیم مناسب از عملکرد میتوکندری ها برای بهینه سازی تولید ATP برای پاسخگویی به نیازهای سلولی ضروری است، در حالی که همزمان به حداقل رساندن اثرات بالقوه مضر تولید رادیکال های آزاد بیش از حد است. یک نیاز حیاتی برای مدولاسیون خوبی از عملکرد میتوکندری، ظرفیت میتوکندری است که به طور مستقل به عنوان دستگاه های بیوشیمیایی عمل می کند و به عنوان بخشی از یک شبکه گسترده و واکنش پذیر است.
مورفولوژی و عملکرد شبکه های میتوکندری توسط تعادل تنظیم شده بین تقسیم و همجوشی تعیین می شود. تقسیم میتوکندری برای ارث بری سلول های دختر میتوکندری در حین تقسیم سلولی [28] و همچنین برای تخریب اتفاجی انتخابی میتوکندری دپولاریزه یا آسیب دیده، نامیده می شود [1]. در مقابل، همجوشی برای تکمیل ژنوم های میتوکندری و به اشتراک گذاری اجزای زنجیره ای انتقال الکترون بین میتوكندری های همسایه [54] ضروری است. در سطح مولکولی، تقسیم و همجوشی میتوکندری توسط GTPases بزرگ مانند دینام تنظیم می شود. سه آنزیم تنظیم کننده فیوژن هستند: Mitofusins 1 و 2 (Mfn1 / 2) پروتئین غشایی بیرونی دو طرفه هستند که متمایز شدن غشای بیرونی با همکاری های heterotypic میان میتوکندری های مجاور [15]، [25]، [37] است، در حالی که OPA1 یک درون پروتئین غشایی که به طور همزمان باعث تطبیق ماتریکس می شود با تنظیم مبرد غشاء درونی [5]. فعالیت GTPase از هر سه پروتئین برای فیوژن قوی [5]، [18] و OPA1 بیشتر توسط پروتئولیز پیچیده در غشای داخلی میتوکندری توسط پروتئازهای OMA1 [14]، PARL [6] و YME1L [45 ] مهم این است که پتانسیل غشایی میتوکندری بدون در نظر گرفتن همجوشی کارآمد برای جلوگیری از ادغام میتوکندری آسیب دیده و سالمی [26] مورد نیاز است.
تقسیم میتوکندری عمدتا توسط یک پروتئین سیتوزول به نام پروتئین 1 وابسته به دینین (Drp1 / DNM1L) کاتالیز می شود. Drp1 از سیتوزول به مکان های آینده نابودی تقسیم شده در غشای بیرونی میتوکندری [43] استخدام می شود. گیرنده های اصلی Drp1 در غشای خارجی فاکتور تقسیم میتوکندری (MF) [32] و، به میزان کم، Fission 1 (Fis1) [51] می باشد. علاوه بر این، یک گیرنده کادویی، MIEF1 / MiD51 کشف شد که فعالیت بیشتری را برای پروتئین Drp1 در مناطق شکافت پالسی [58] محدود می کند. هنگامی که در غشای بیرونی میتوکندری قرار می گیرد، Drp1 oligomerizes به ساختار مارپیچی در اطراف بدن از میتوکندریون و سپس با استفاده از انرژی حاصل از هیدرولیز GTP برای میانجیاد زیست فیزیکی غشاهای بیرونی و داخلی میتوکندری [17]. لوله های مشتق شده از رتیکولوم اندوپلاسمی به عنوان یک تداخل اولیه از میتوکندری قبل از الگومیزاسيون Drp1 عمل می کنند، که نشان می دهد که میتوکندری های غیرمتعارف گسترده تر از محدوده مجاز یک مارپیچ Drp1 [12] است. دینامیک آتیین نیز برای تعاملات میتوکندری ER که قبل از تقسیم میتوکندری [24] مهم است. Drp1 علاوه بر نقش آن در تقسیم میتوکندری، تقسیم پراکسسیوم ها [40] را تجزیه می کند.
Drp1 بسیار شبیه به پروتئین دینامانی شناخته شده است که در هر دو پروتئین حاوی دامنه GTPase N-terminal است، دامنه میانی که برای خود الگومریزیزه حیاتی است و دامنه GFTase [31] C-terminal G-terminal است. Drp1 انتخابی برای غشاهای میتوکندری را از طریق ترکیبی از تعاملات با پروتئین گیرنده Mff و Fis1 و همچنین از طریق وابستگی آن به قلب یولپین فسفولیپید خاص با میتوکندری از طریق حوزه ی منحصر به فرد B-Drp1 [2]، به دست می دهد. Drp1 معمولا به عنوان یک هوموتوترامر در سیتوپلاسم وجود دارد، و مونتاژ بالاتر در مکان های تقسیم میتوکندری توسط قلمرو مرکزی Drp1 [3] متصل می شود.
با توجه به ارتباط ضمنی بین عملکرد میتوکندری و مسیر KEAP1-Nrf2-ARE، اثرات فعال سازی Nrf2 بر ساختار و عملکرد میتوکندری بررسی شد. ما در اینجا نشان می دهیم که SFN موجب افزایش فشار خون میتوکندری می شود که به طور غیر منتظره مستقل از هر دو Nrf2 و KEAP1 است. این اثر SFN از طریق مهار عملکرد Drp1 است. ما بیشتر نشان می دهیم که SFN مقاومت در برابر آپوپتوز را به وجود می آورد که مستقل از Nrf2 است و تقلید آن در سلول هایی که از Drp1 دیده می شوند، مشاهده می شود. این داده ها به طور کلی نشان می دهد که علاوه بر پاکسازی و فعال کردن Nrf2، SFN مولکولی پویایی میتوکندری را حفظ می کند و حفظ تناسب و بقاء سلولی را حفظ می کند.
نتایج
سولفورفان موجب بروز Hyperfusion مستقل از Mitohondria و Nrf2 / KEAP1 می شود.
در طی مطالعه تأثیرات فعال سازی Nrf2 بر پویایی شبکه میتوکندری ، متوجه شدیم که درمان سلول های اپیتلیال رنگدانه شبکیه چشم انسان (RPE-1) جاودانه با سولفورافان (SFN) ، یک فعال کننده قوی سیگنالینگ Nrf2 ، باعث همجوشی قوی شبکه میتوکندری وقتی با سلولهای کنترل شده با وسیله نقلیه مقایسه شود (شکل 1 A و B). مورفولوژی میتوکندری در این سلول ها تا حد زیادی شبیه میتوکندری در سلولهای تخلیه شده توسط siRNA از Drp1 درون زا ، عامل اصلی شکافت میتوکندری است (شکل 1 A). این نتیجه این ایده جذاب را ایجاد می کند که شکافت میتوکندری و وضعیت همجوشی مستقیماً به سطح Nrf2 در سلول پاسخ می دهد. با این حال ، تحریک سلول ها با سایر تثبیت کننده ها و فعال کننده های Nrf2 مانند مهارکننده پروتئازوم MG132 ، tBHQ پرو اکسیدان یا ناکارد مهارکننده Nrf2 KEAP1 باعث همجوشی میتوکندری (شکل 1 A و B) نمی شود. تثبیت Nrf2 توسط این دستکاری ها توسط وسترن بلات برای Nrf2 درون زا تأیید شد (شکل 1C). علاوه بر این ، بیان Nrf2 برای همجوشی میتوکندری ناشی از SFN قابل توزیع بود ، زیرا شکست Nrf2 درون زا با siRNA برای مقابله با این فنوتیپ شکست خورد (شکل 1D F). از آنجا که SFN با اصلاح کووالانسی باقیمانده های سیستئین KEAP1 [2] مسیر KEAP1-Nrf21-ARE را تحریک می کند ، ما KEAP1 را پایین آوردیم تا ببینیم آیا بیش از حد میتوکندری ناشی از SFN از طریق یک مسیر مستقل از KEAP1 وابسته به KEAP2 تحریک می شود. با این حال ، کاهش KEAP1 همچنین نتوانست همجوشی میتوکندری ناشی از SFN را لغو کند (شکل 1G I). در حقیقت ، SFN شکل گیری پیش شکافت ناشی از کاهش KEAP1 را معکوس کرد (شکل 1G ، صفحه b در مقابل صفحه d). این نتایج نشان می دهد که درمان SFN باعث همجوشی میتوکندری مستقل از مسیر متعارف KEAP1-Nrf2-ARE می شود و ما را به بازجویی سوق می دهد که آیا SFN به طور مستقیم بر اجزای شکافت میتوکندری یا ماشین آلات همجوشی تأثیر می گذارد.
شکل 1 SFN منجر به همجوشی میتوکندری مستقل Nrf2 / KEAP1 می شود. (A) سلولهای RPE-1 با siRNA های مشخص شده ترانسفکت شدند و 3 روز بعد با DMSO یا فعال کننده های Nrf2 SFN (50؟ M) ، MG132 (10؟ M) یا tBHQ (100؟ M) به مدت 4 ساعت تحت درمان قرار گرفتند. میتوکندری (قرمز) با آنتی بادی ضد Tom20 برچسب گذاری شده و هسته ها (آبی) با DAPI ضد رنگ می شوند. (B) نمودار نشان دادن کمی از نمره گذاری مورفولوژی میتوکندری از (A). > 50 سلول در هر شرایط به روشی کور ارزیابی شد. (C) نماینده وسترن بلات از (A). (D) سلولهای RPE-1 با 10 نانومتر siRNA ترانسفکت شدند و 3 روز بعد با SFN به مدت 4 ساعت قبل از ثابت شدن و رنگ آمیزی مانند (A) تحت درمان قرار گرفتند. (E) نمودار نشان دهنده کمی سازی نمره فنوتیپ میتوکندری از (D) است. > 100 سلول در هر شرایط به روشی کور ارزیابی شد. (F) وسترن بلات نماینده از (D). (G) سلولها مانند (D) با siCON یا siKEAP1 ترانسفکت و تحت درمان قرار گرفتند. سلولهای (H) از (G) براساس (B) و (E) بر اساس مورفولوژی میتوکندری امتیازدهی شدند. (I) نماینده وسترن بلات از (G). داده های موجود در (B) ، (E) و (H) هر یک از 3 آزمایش مستقل جمع آوری شد و اهمیت آماری توسط آزمون t دو دانش آموزی تعیین شد. میله های خطا +/- SD را منعکس می کنند (برای تفسیر منابع در این افسانه شکل ، خواننده به نسخه وب این مقاله ارجاع می شود).
سولفورفان باعث آسیب رساندن به انجمن میتوکندری Drp1 می شود
بر اساس یافته هایی که درمان SFN موجب هیپوفیز کردن میتوكندری می شود، ما تصور كردیم كه این فنوتیپ یا نتیجه فعالیت فیوژن بیش از حد یا مهار فعالیت های تجزیه است. برای تمایز بین این دو ویژگی، ما مورفولوژی پراکسیزوم را در حضور و عدم وجود SFN مقایسه کردیم. پراکسیزم ها شبیه به میتوکندری هستند، زیرا آنها اندام های پویا هستند که طول آنها به طور مداوم در شار [44] می باشد. پراکسیزوم ها حاوی Fis1 و MF در غشای خارجی هستند و در نتیجه اهداف شبیه سازی [1]، [22] Drp23 می باشند. با این حال، پراکسسیوم ها از ماشین آلات همجوشی شبکه های میتوکندری استفاده نمی کنند و در نتیجه فوجی [39] را انجام نمی دهند. در عوض، تقسیم پراکسسیوم با افزایش پراکسیزوم های موجود از طریق افزودن غشای غشاء و پروتئین ها [44] مخالف است. از آنجا که پراکسیوم ها Mfn1 / 2 و OPA1 ندارند، ما تصور کردیم که اگر SFN به جای مهار ماشین آلات تجزیه کننده، ماشین آلات فیوز را فعال کند، طول پراکسیوم موثر نخواهد بود. در سلول های تحت درمان با وسیله نقلیه، پراکسیزوم ها به عنوان اندام کوتاه، دور، پانکستیوم (شکل 2، پانل های b و d) نگهداری می شوند. با این حال، درمان با SFN طول پروکسیوم را با 2 برابر با سلول های کنترل (شکل 2، پانل های f و h) افزایش داد. علاوه بر این، بسیاری از پراکسیوم ها در نزدیکی مرکز منقبض شده اند، که نشان دهنده نقص انکساری بالقوه است (شکل 2، پانل h، سرها). به همین ترتیب، پراکسیزم ها در سلول های انتقال دهنده siRNA Drp1 غیر طبیعی طولانی بودند (شکل 2، پانل ها j و l)، تایید کرد که Drp1 برای تقسیم پراکسسیوم مورد نیاز است و پیشنهاد می کند که درمان SFN باعث ایجاد فنوتیپ های میتوکندریال و پراکسسیوم می شود با اختلال در دستگاه های تجزیه کننده.
شکل 2 SFN باعث افزایش طول پراکسیوم می شود. (A) سلولهای RPE-1 با 10 نانومولار نشان داده شده از siRNA و 3 روز بعد با DMSO یا 50؟ M SFN به مدت 4 ساعت تحت درمان قرار گرفتند. پراكسی زومها (سبز) با آنتی بادی ضد PMP70 ، میتوكندری با MitoTracker (قرمز) و DNA با DAPI رنگ آمیزی شدند. حفره های بزرگ پراكسیزوم در سمت راست (تابلوهای d ، h و l) نشان داده شده اند تا تجسم تغییرات در مورفولوژی ناشی از تخلیه SFN و Drp1 را تسهیل كنند. نوک پیکان نقاط انقباض را برجسته می کند. (برای تفسیر منابع در این افسانه شکل ، خواننده به نسخه وب این مقاله ارجاع می شود).
سپس تعیین کردیم که چگونه SFN عملکرد Drp1 را محدود می کند. احتمالات شامل کاهش در سطوح بیان، استخدام / حفظ در میتوکندری، الیگومریزاسیون، یا فعالیت آنزیمی GTPase بود. نقص در هر یک از اینها منجر به کاهش شکافت و هیپرفیوژن میتوکندری می شود. ما تغییرات قابل تکرار در سطوح پروتئین Drp1 را پس از درمان با SFN شناسایی نکردیم (شکل 1C و 3A)، و بنابراین به این نتیجه رسیدیم که SFN پایداری یا بیان Drp1 را تغییر نمیدهد، مطابق با نیمه عمر Drp1 بیش از 10 ساعت [50]. و درمان SFN ما مدت زمان کمتری دارد. در مرحله بعد، بررسی کردیم که آیا SFN بر جذب یا حفظ Drp1 در میتوکندری تأثیر می گذارد یا خیر. مطالعات شکنش نشان داد که SFN باعث از دست دادن Drp1 از بخش میتوکندری می شود (شکل 3A، خطوط 7-8 و شکل 3B). همانطور که قبلاً گزارش شد [43]، تنها بخش کوچکی از Drp1 (3٪) با شبکه میتوکندری در هر زمان معین در شرایط حالت پایدار همراه است و بیشتر آنزیم در سیتوپلاسم ساکن است (شکل 3A، خطوط 5-8). ). این دادههای تقسیمبندی با استفاده از تجزیه و تحلیل هممحلسازی تأیید شد که کاهش 40 درصدی را در کانونهای Drp1 نقطهگذاری شده با میتوکندری پس از درمان SFN نشان داد (شکل 3C و D). با هم، این داده ها نشان می دهد که همجوشی میتوکندری ناشی از SFN، حداقل تا حدی، به دلیل ارتباط ضعیف Drp1 با میتوکندری است. دادههای ما بین اینکه آیا SFN با جذب میتوکندریایی در مقابل حفظ میتوکندری Drp1 تداخل میکند یا هر دو، تمایز قائل نمیشود، زیرا تجزیه و تحلیل Drp1 درونزا برای تجسم GTPase توسط میکروسکوپ سلول زنده قابل قبول نبود.
شکل 3 SFN باعث از بین رفتن Drp1 از میتوکندری می شود. (A) تقسیم سلول های سلولی RPE-1 پس از 4 ساعت از DMSO یا SFN. با استفاده از SDS-PAGE، لسیاتهای کل سلولی (WCL)، هسته ای (Nuc)، سیتوزول (Cyto) و میتوکندری خام (Mito) حل شده و برای آنتی بادی های غلط غربی مورد استفاده قرار می گیرند. مهاجرت نشانگرهای مولکولی در سمت چپ نشان داده شده است. (B) نمودارها نشان دهنده اندازه گیری densitometry Drp1 در بخش های نشان داده شده از (A) است. (C) سلول های RPE-1 با 10 nM siCON یا siDrp1 و 3 روز بعد با DMSO یا SFN برای 4 h تجویز شدند. Drp1 (سبز) با یک آنتی بادی anti-Drp1، میتوکندری با MitoTracker (قرمز) و هسته ای با DAPI (آبی) تجسم شد. (D) تجزیه و تحلیل خودکار محلی سازی Drp1 و سیگنال MitoTracker از (C). داده ها در (B) و (D) به ترتیب از آزمایشات مستقل 3 و 5 جمع آوری شده و از نظر آماری معنیداری با آزمون t-student دو طرفه بود. نوارهای خطا +/- SD را نشان می دهند و ستاره ها اهمیت آماری را نشان می دهند. (برای تفسیر ارجاع به رنگ در این رقم افسانه، خواننده به نسخه وب این مقاله ارجاع داده می شود).
سولفورفان محافظتی علیه آپوپتوز ناشی از استاستوسپورتین را مستقل از Nrf2
کار قبلی نشان داده است که شکافت میتوکندری در تشکیل منافذ در غشای خارجی میتوکندری که توسط Bax/Bak در طول آپوپتوز ایجاد میشود، مجاز است [11]. نشان داده شده است که Drp1 به طور انتخابی در طول آپوپتوز به میتوکندری ها وارد می شود [11] و مطابق با این، میتوکندری های تکه تکه شده در اوایل فرآیند مشاهده شده اند [27]. برعکس، تصور می شود که مهار شکافت میتوکندری با مسدود کردن تشکیل منافذ غشای خارجی که امکان آزادسازی سیتوکروم c را فراهم می کند، آپوپتوز را مهار می کند [53]. بر این اساس، تحریک همجوشی میتوکندری پیشرفت آپوپتوز ناشی از ترکیباتی از جمله استاوروسپورین (STS) را به تاخیر می اندازد [14]. برای تعیین اینکه آیا SFN از سلولهای RPE-1 در برابر آپوپتوز با واسطه STS محافظت میکند و اگر چنین است، آیا این به Nrf2 نیاز دارد یا خیر، ما سنجشی را برای القای آسان برش پلی مراز پلی مراز ADP ریبوز (PARP)، بستری از کاسپاز-3 فعال شده و نشانگر قطعی ایجاد کردیم. آپوپتوز درمان سلولهای RPE-1 با 1 میکرومولار STS بهمدت 6 ساعت تنها باعث برش بسیار کم PARP شد، اما با درمان مشترک SFN از این امر جلوگیری شد (به عنوان مثال، شکل 4A، خط 3 در مقابل 4). برای افزایش استحکام این روش، ما سلولها را با پیشدرمان کردن سلولها با siRNA که فاکتور ضد آپوپتوز Bcl-XL را هدف قرار میدهد، به آپوپتوز ناشی از STS بیشتر حساس کردیم. این پیش درمانی بیان Bcl-XL را کاهش داد و به طور قابل توجهی برش PARP را به عنوان تابعی از زمان قرار گرفتن در معرض STS ارتقا داد (شکل 4B، خط 2 را با خطوط 4-10 مقایسه کنید). نکته مهم این است که 2 ساعت قبل از درمان با SFN برش PARP را در سلول های در معرض STS کاهش داد (شکل 4C، خط 3 در مقابل 4 و خط 5 در مقابل 6). به همین ترتیب، سلول هایی که به طور پایدار از Nrf2 توسط CRISPR/Cas9 تخلیه شده بودند، به طور قابل قیاسی از سمیت STS توسط پیش درمان SFN محافظت شدند (شکل 4C، خط 11 در مقابل 12 و خط 13 در مقابل 14 و شکل 4D). این حفاظت با استفاده از برش PARP (شکل 4C و D) و مورفولوژی سلولی (شکل 4E) به عنوان بازخوانی مشاهده شد. کارایی کاهش Nrf2 توسط CRISPR/Cas9 توسط وسترن بلات تایید شد (شکل 4C، بلات Nrf2). همانطور که پیشبینی میشد، سلولهای کاهشدهنده Drp1، که همچنین یک فنوتیپ هیپرفیوژن (شکل 1A) را ایجاد میکند، در مقایسه با سلولهای کنترل انکوبهشده با SFN (شکل 4F و G)، برش PARP را در پاسخ به STS مسدود کرد. با هم، این یافتهها با SFN که از طریق ظرفیت آن برای محدود کردن عملکرد Drp1، مستقل از تثبیت و فعالسازی Nrf2، محافظت در برابر آپوپتوز ایجاد میکند، سازگار است.
شکل 4 اثرات محافظت از سلول SFN مستقل از بیان Nrf2 (A) سلولهای RPE-1 قبل از درمان با DMSO ، 50 استارواسپورین 2 M یا ST 1 یا 50A از قبل با DMSO یا 6؟ M SFN تحت درمان قرار گرفتند. etoposide M به مدت XNUMX ساعت و برای ضد وسترن بلات پردازش شدند. (B) سلولهای RPE-1 با 2.5 نانومتر siCON ، 1 نانومتر siBcl-XL یا 2.5 نانومول siBcl-XL ترانسفکت شدند و 3 روز بعد با DMSO یا 1؟ M STS به مدت 2 ، 4 یا 6 ساعت تحت درمان قرار گرفتند. لکه های وسترن نماینده نشان داده شده و انتقال نشانگرهای وزن مولکولی در سمت چپ نشان داده شده است. (C) CRISPR / Cas9 تولید شده از نوع وحشی (Nrf2WT) و Nrf2 حذفی (Nrf2KO) سلولهای RPE-1 با 1nM siBcl-XL ترانسفکت شدند و 3 روز بعد با DMSO یا 50؟ M SFN به مدت 2 ساعت تحت درمان قرار گرفتند . پس از آن ، سلول ها با 1؟ M STS به مدت 2 ، 4 یا 6 ساعت تحت درمان قرار گرفتند. لکه های وسترن نماینده با آنتی بادی های نشان داده شده نشان داده شده است. (D) مقدار PARP خرد شده به عنوان درصدی از PARP کل (شکافدار + شکافته نشده) از 3 آزمایش مستقل. نکته مهم ، سطح PARP شکاف خورده است که آیا سلولها Nrf2 را بیان می کنند یا نه ، نشان می دهد که محافظت SFN از STS مستقل از فاکتور رونویسی است. (E) تصاویر کنتراست فاز 20X بلافاصله قبل از برداشت مواد لیزاتی از (C) گرفته شده است. نوار مقیاس = 65 m. (F) وسترن بلات نمایندگی نشان می دهد که کاهش Drp1 محافظت تقریباً قابل مقایسه ای را از STS به عنوان درمان SFN ایجاد می کند. سلولهای RPE-1 با 1 نانومتر siBcl-XL ترانسفکت شدند و علاوه بر این با 10 نانومتر siCON یا 10 نانومتر siDrp1 ترانسفکت شدند. 3 روز بعد ، سلولهای siCON همانند (A) و (C) با SFN تحت درمان قرار گرفتند و سپس 4 ساعت در معرض STS قرار گرفتند تا قبل از برداشت و پردازش برای وسترن بلات با آنتی بادی های مشخص شده. (G) همان (D) برای داده های ارائه شده در (F) که از 3 آزمایش مستقل گردآوری شده است. میله های خطا +/- SEM را منعکس می کنند
بحث
ما کشف کرده ایم که SFN مدولاسیون پویایی تقسیم / همجوشی میتوکندری مستقل از اثرات آن بر مسیر KEAP1-Nrf2-ARE است. این امر به دلیل یک پیوند فرض شده بین اختلال عملکرد میتوکندریال و تولید ROS و ضرورت استحکام رادیکال های آزاد ماتوخناندی از طریق فعال شدن Nof2 جالب است. این تاثیر کاربردی اضافی SFN از اهمیت بالایی برخوردار است زیرا بیشتر از آزمایشات بالینی 30 در حال انجام آزمایش SFN برای درمان انواع بیماری ها از جمله سرطان پروستات، بیماری های انسدادی ریوی و بیماری سلول های سگ [7]، [10]، [ 47]
از آنجا که SFN isothiocyanate [56] است و آن سیگنال های انتقالی KEAP2 را به طور مستقیم آکیل کردن سیستین های بحرانی KEAP1 برای مهار توزیع NLF2 [21] فعال می کند، بدین معنی است که SFN اثرات پرولوژی خود را بوسیله تعدیل فعالیت یک عامل تقسیم یا فیوژن از طریق اصلاح سیستین . داده های ما به شدت از Drp1 حمایت می کند که توسط SFN منفی تنظیم می شود، هرچند که آیا GTPase یک هدف مستقیم آسیل سازی است که هنوز مشخص نشده است. علیرغم این شکاف دانش، عملکرد Drp1 به صراحت توسط SFN به خطر افتاده است؛ چرا که هر دو میتوكندری و پراكسیوم ها در پاسخ به درمان SFN بیش از حد فرسوده می شوند و این ارگانل ها Drp1 را برای رویدادهای مربوط به تجزیه آن [38] به اشتراک می گذارند. علاوه بر این، SFN میزان Drp1 را کاهش می دهد که در میتوکندری موضعی و تجمع می یابد (شکل. 3). از آنجایی که آزمایشات ما با تمام پروتئین های درونی انجام می شود، تشخیص Drp1 در محل های تقسیم میتوکندری تحت شرایط پایدار است و در نتیجه ما نمی توانیم بین استخدام و نقص نگهداری از آنزیم ناشی از SFN را تشخیص دهیم. علاوه بر این، ما نمی توانیم احتمال این که SFN یک گیرنده در میتوکندری (Fis1 یا MF) آشکار می کند تا مانع جذب Drp1 شود، ما معتقدیم Drp1 به طور مستقیم اصلاح می شود. Drp1 داراي 9 سيستئين است که هشت نفر از آنها در دامنه مياني قرار دارند که براي اليگومريزاسيون لازم است [3] و يکي از آنها در دامنه افترتور GTPase (GED) در انتهاي C Drp1 قرار دارد. Acylation مستقیم هر کدام از این سيستئین ها می تواند باعث اختلال فعالیت در Drp1 شود و بنابراین اثر SFN بر پویایی میتوکندری را تحت تأثیر قرار می دهد. به طور قابل توجهی، کار قبلی نشان می دهد که نقص در oligomerization و فعالیت کاتالیزوری می تواند حفظ Drp1 در میتوکندری [52]. Cys644 در دامنه GED هدف خاصی است که بر اساس کار قبلی نشان می دهد که جهش های جهش های این فنوتیپ های سیتئین که فعالیت Drp1 GTPase [4] را مختل می کنند و این سیتستین خاص با الکتروفیل های واکنشی تیول [9] اصلاح می شود. رفع این سوال فوقالذکر نیاز به اعتبار سنجی طیف سنجی دارد. به طور خلاصه، ما یک تابع جدید، سیتوپروتیک را برای SFN ترکیبی بالینی داریم. علاوه بر فعال شدن عامل اصلی رونویسی ضد عفونی کننده Nrf2، SFN باعث افزایش تلفات میتوکندری و پراکسسیوم می شود و این اثر مستقل از Nrf2 می باشد. مکانیسم اصلی این پدیده شامل کاهش عملکرد GTPase Drp1، واسطه اصلی تقسیم میتوکندری و پراکسسیوم است. یک نتیجه عمده از SFN-متمرکز فیوژن میتوکندری این است که سلول ها به اثرات سمی استوروسورپین القا کننده آپوپتوز مقاوم می شوند. این اثر اضافی سیتوپروتئینی SFN میتواند از ابزار بالینی خاصی در بیماریهای نورودژنراتیک متعدد باشد که سن آنها عامل اصلی خطر است (به عنوان مثال بیماری پارکینسون، بیماری آلزایمر، دژنراسیون ماکولای مربوط به سن) به عنوان این بیماری با آپوپتوز همراه است و کاهش سطوح و / یا اختلال تنظیم Nrf2 [35]، [36]، [48].
مواد و روش ها
تست آپوپتوز
همانطور که در زیر نشان داده شده ، سلولها بذر و با siRNA ترانسفکت شدند سلول ها برای القای همجوشی میتوکندری به مدت 50 ساعت با 2μ M سولفورافان تحت درمان قرار گرفتند و سپس با القای 1 استاروسپورین M برای القای آپوپتوز تحت درمان قرار گرفتند. در زمان برداشت ، محیط در لوله های جداگانه جمع آوری شد و تحت سلولهای سانتریفوژ با سرعت بالا به سلولهای آپوپتوز گلوله قرار گرفت. این گلوله سلولی با سلولهای چسبنده ترکیب شده و در بافر 2 برابر غلیظ Laemmli حل شد. نمونه ها تحت لکه گیری ضد PARP قرار گرفتند.
CRISPR / Cas9 Construct Generation
برای ایجاد LentiCRISPR / eCas9 1.1، LentiCRISPR v2 (addgene #52961) ابتدا با Age1 و BamH1 بریده شد. سپس، SpCas9 از eSpCas9 1.1 (addgene #71814) PCR amplified با استفاده از primer ها در رده های Age1 و BamH1 با استفاده از پرایمر های زیر (جلو AGCGCACCGGTTCTAGAGCGCTGCCACCATGGACTATAAGGACCACGAC، معکوس AAGCGCGGATCCCTTTTTCTTTTTTGCCTGGCCGG) و در برش برش بالا بالا لیگاد شده است. توالی sgRNA با استفاده از Benchling.com تعیین شد. پارامترها برای دنبال کردن توالی کدگذاری با بالاترین میزان بر روی هدف و کمترین نمرات غیر هدف قرار گرفتند. توالی های زیر (با هدف قرار دادن توالی زیر خط دار، HS sgNFE2L2 # 1 حس CACCGCGACGGAAAGAGTATGAGC، آنتی AAACGCTCATACTCTTTCCGTCGC؛ HS sgNFE2L2 # 2 CACCGGTTTCTGACTGGATGTGCT معنا، AAACAGCACATCCAGTCAGAAACC آنتی سنس؛ HS sgNFE2L2 # 3 حس CACCGGAGTAGTTGGCAGATCCAC، آنتی AAACGTGGATCTGCCAACTACTCC) آنیل و مسدود به BsmB1 قطع LentiCRISPR / eCas9 1.1 شد. سلول های RPE-1 آلوده به لنفوویروس با پوموویسیین انتخاب شدند و به عنوان یک جمعیت جمع شده نگهداری شدند. ناکوته توسط ایمونوفلورسانس و غربالگری غربی تأیید شد.
فرهنگ سلولی و ترانسفکشن
سلولهای اپیتلیال رنگدانه ای شبکیه انسان که با تلومراز (RPE-1) تبدیل شده اند (ATCC) در اصلاح شده عقاب متوسط Dulbecco (DMEM) حاوی 1 گرم در لیتر گلوکز همراه با پنی سیلین ، استرپتومایسین ، 1X کوکتل اسید آمینه غیر ضروری (Life Technologies) ، و 10٪ سرم جنین گاو (فن آوری های زندگی). برای انتقال siRNA ، 30,000 cells 35,000 سلول در میلی لیتر یک شب بذر شد. سلولها 10 نانومولار siRNA رقیق شده در DMEM بدون سرم و همراه با 0.3٪ واکنش ترانسفورین اینترفرین (PolyPlus) دریافت کردند. برای حساسیت به آپوپتوز ، سلول ها 1 nM Bcl-XL siRNA دریافت کردند. سلول ها 2 3 روز پس از عفونت برداشت شدند.
مواد شیمیایی، آنتی بادی، و siRNA Oligos
آنتی بادی های ضد؟ توبولین (سیگنالینگ سلولی) ، بتوبولین (سیگما) ، Drp1 (BD Biosciences) ، KEAP1 (پروتئین تک) ، لامین B1 (آبکم) ، PARP (سیگنالینگ سلولی) ، PMP70 (آبکم) و Tom20 (BD علوم زیستی) ) در رقت 1: 1000 برای وسترن بلات و برای ایمونوفلورسانس استفاده شد. از آنتی بادی خرگوش ضد Nrf2 در خانه 1: 2000 برای وسترن بلات استفاده شد [34] ، [59]. از سولفورافان (سیگما) و استاوروسپورین (توکریس) به ترتیب در 50؟ M و 1؟ M استفاده شد. siRNA ها در برابر Drp1 (Dharmacon) ، Nrf2 (Dharmacon) ، KEAP1 (سیگنالینگ سلول) و Bcl-XL (سیگنالینگ سلولی) در 10 نانومتر استفاده شد مگر اینکه خلاف آن ذکر شده باشد.
ایمونوفلورسانس و در برچسب Vivo
سلول های بذر شده روی روکش های شیشه ای 18 میلی متری با وسیله نقلیه یا دارو تحت تیمار قرار گرفتند ، در 3.7٪ فرمالدئید فیکس شدند و سپس در 0.2٪ تریتون X-100 / PBS روی یخ به مدت 10 دقیقه نفوذ یافتند. آنتی بادی های اولیه در 3 album آلبومین سرم گاو (BSA) در PBS یک شب در دمای 4 درجه سانتیگراد انکوبه شدند. به دنبال شستشوی PBS ، سلول ها به مدت 1 ساعت در آنتی بادی های ثانویه مزدوج مناسب الکسا 488- یا الکسا 546- ، رقت 1: 1000) و 0.1 گرم در میلی لیتر DAPI (سیگما) در 3٪ BSA / PBS انکوبه شدند. میتوکندری یا توسط ایمونوفلورسانس ضد Tom20 یا با سلول های جوجه کشی در 200 نانومولار MitoTracker Red CMXRos (پروب مولکولی ، شرکت) در DMEM بدون سرم به مدت 30 دقیقه در 37 درجه سانتیگراد قبل از تثبیت مشاهده شد.
میکروسکوپ و تحلیل تصویر
نمونه های ایمونوفلورسانس بر روی میکروسکوپ کانفوکال LSM710 (Carl Zeiss) مشاهده شد. میکروگرافها با استفاده از اهداف غوطه وری روغن 63X یا 100X و تصاویر با استفاده از Adobe Photoshop CS6 تنظیم و بهبود یافته اند. تجزیه و تحلیل محلی سازی با استفاده از Carl Zeiss LSM710 ویژگی محلی سازی مشترک با آستانه تنظیم دستی انجام شد در حالی که کور به هویت نمونه ها. میله های مقیاس در کل ، مگر اینکه خلاف آن مشخص شده باشد ، 10 می باشد. مورفولوژی میتوکندری با نمره گذاری کور ارزیابی شد. اگر میتوکندری سلول به صورت چند نقطه ای ، گرد و تفکیک پذیر حفظ شود ، سلول به عنوان "شکافت" امتیاز گرفت. اگر میتوکندری های فردی قابل تشخیص نبودند و کل شبکه میتوکندری به طور مداوم ظاهر می شد ، سلول به عنوان "فیوژن" به ثمر رسید. تمام سلولهای دیگر ، از جمله سلولهای دارای میتوکندری خوشه ای ، به عنوان "متوسط" امتیاز بندی شدند.
تقسیم بندی های زیر سلولی
سلولهای RPE-1 تا محل تلاقی رشد کردند. پس از شستشوی PBS ، سلول ها در دمای 600 گرم به مدت 10 دقیقه تحت سانتریفیوژ قرار گرفتند و در بافر جداسازی 600 ل L (210 میلی متر مانیتول ، 70 میلی متر ساکارز ، 5 میلی متر MOPS ، 1 میلی متر EDTA pH 7.4 + 1 میلی متر PMSF) مجدداً تعلیق شدند. سیستم تعلیق 30 بار در هموژنایزر دانس لیز شد. کسری از هموژن به عنوان یک لیزات سلول کامل حفظ شد. باقیمانده در 800 گرم به مدت 10 دقیقه به هسته گلوله در سانتریفوژ قرار گرفت. سوپرونتنت ها برای پاک کردن هسته های باقیمانده و سلول های بدون لیزر تحت حرارت 1500 گرم به مدت 10 دقیقه تحت سانتریفوژ قرار گرفتند. این ماده رویی در 15,000 گرم برای 15 دقیقه به میتوکندری گلوله در سانتریفیوژ قرار گرفت. مایع رویی به عنوان "کسر سیتوزولی" حفظ شد. گلوله را به آرامی با PBS شستشو داده و در بافر جداسازی مجدداً معلق نمود. غلظت پروتئین هر بخش با استفاده از روش بیسینکونینیک اسید (BCA) اندازه گیری شد و مقادیر معادل پروتئین توسط SDS-PAGE حل شد.
Blotting غرب
سلول ها در PBS شسته شدند و در بافر حل کننده 2 بار غلیظ Laemmli (100 میلی مولار Tris [pH 6.8]، 2٪ SDS، 0.008٪ بروموفنول بلو، 2٪ 2- مرکاپتواتانول، 26.3٪ گلیسرول، و 0.001٪ Pyrinin) حل شدند. لیزها قبل از بارگذاری روی ژل پلی آکریل آمید سدیم دودسیل سولفات (SDS) به مدت 5 دقیقه جوشانده شدند. پروتئین ها به غشاهای نیتروسلولزی منتقل شدند و غشاها به مدت 1 ساعت در شیر/TBST 5 درصد مسدود شدند. آنتی بادی های اولیه در شیر/TBST 5 درصد رقیق شده و با بلات یک شبه در دمای 4 درجه سانتیگراد انکوبه شدند. آنتی بادی های ثانویه کونژوگه ترب پراکسیداز (HRP) در شیر/TBST 5 درصد رقیق شدند. بلات با نورتابی شیمیایی افزایش یافته پردازش شد و کمی سازی چگالی سنجی با استفاده از نرم افزار ImageJ انجام شد.
سولفورفان یک ماده شیمیایی است که از مجموعه ایزوتیوسیانات مواد تشکیل دهنده سولفوریک به دست آمده از سبزیجات cruciferous، از جمله بروکلی، کلم، گل کلم، کلم و کلث ها، از جمله دیگر است. سولفورفان تولید می شود زمانی که آنزیم میروزیناز، گلوکوراپامین، گلوکوزینولات، را به سولفورفان تبدیل می کند که همچنین به عنوان سولفورفان گلوکوزینولات شناخته می شود. جوانه های بروکلی و گل کلر دارای بالاترین غلظت گلوکوراپامین یا پیش ماده سولفورفان هستند. تحقیقات انجام شده نشان داده است که سولفورفان توانایی های آنتی اکسیدانی بدن انسان را برای جلوگیری از مسائل مختلف بهداشتی افزایش می دهد. دکتر الکس جیمنز DC، CCST Insight
سولفورافان و تأثیرات آن بر سرطان ، مرگ و میر ، پیری ، مغز و رفتار ، بیماری های قلبی و موارد دیگر
ایزوتیوسیانات ها یکی از مهمترین ترکیبات گیاهی هستند که می توانید در رژیم غذایی خود داشته باشید. در این ویدئو من جامع ترین مورد برای آنهایی است که تاکنون ساخته شده است. فاصله کم توجهی؟ با کلیک کردن روی یکی از نقاط زمانی زیر، موضوع مورد علاقه خود را پر کنید. جدول زمانی کامل در زیر
بخش های کلیدی:
00: 01: 14 - سرطان و مرگ و میر
00: 19: 04 - پیری
00: 26: 30 - مغز و رفتار
00: 38: 06 - خلاصه نهایی
00: 40: 27 - دوز
جدول زمانی کامل:
00: 00: 34 - معرفی سولفورفان، تمرکز اصلی این ویدئو است.
00: 01: 14 - مصرف سبزیجات کریستالی و کاهش مرگ و میر در همه موارد.
00: 02: 12 - خطر سرطان پروستات.
00: 02: 23 - خطر سرطان مثانه.
00: 02: 34 - سرطان ریه در افراد سیگاری خطر دارد.
00: 02: 48 - خطر ابتلا به سرطان پستان.
00: 03: 13 - Hypothetical: اگر سرطان دارید، چه؟ (مداخله)
00: 03: 35 - مکانیسم قابل قبول داده های وابسته به سرطان و مرگ و میر.
00: 04: 38 - سولفورفان و سرطان.
00: 05: 32 - شواهد حيواني نشان دهنده اثرات شديد عصاره برانکاري بر رشد تومور مثانه در موش صحرايي.
00: 06: 06 - اثر مکمل مستقیم سولفورفان در بیماران مبتلا به سرطان پروستات.
00: 07: 09 - ذخیره سازی بیولوژیکی متابولیت های ایزوتوسیانات در بافت های پستان واقعی.
00: 08: 32 - مهار سلول های بنیادی سرطان پستان.
00: 08: 53 - درس تاریخ: برنجهای با خواص سلامتی حتی در روم باستان تاسیس شده اند.
00: 09: 16 - توانایی سولفورفان برای افزایش دفع سرطان زایی (بنزن، آکرولئین).
00: 09: 51 - NRF2 به عنوان یک سوئیچ ژنتیکی از طریق عناصر پاسخ آنتی اکسیدان.
00: 10: 10 - چگونه فعالیت NRF2 باعث افزایش دفع سرطانزا از طریق ترکیبات گلوتاتیون-S می شود.
00: 10: 34 - جوانه های بروکسل گلوتاتیون S-transferase را افزایش می دهند و آسیب DNA را کاهش می دهند.
00: 11: 20 - نوشیدنی نای بروکلی باعث افزایش دفع بنزن توسط 61٪ می شود.
00: 13: 31 - هموگلوبین بوته بروکلی آنزیم های آنتی اکسیدان را در فضای باز بالا می برد.
00: 15: 45 - مصرف سبزیجات کریستالی و مرگ و میر بیماری قلبی.
00: 16: 55 - پودر تخم مرغ بروکلی موجب افزایش چربی خون و خطر بیماری کلیوی قلب در افراد مبتلا به دیابت نوع 2 می شود.
00: 19: 04 - شروع فصل پیری.
00: 19: 21 - رژیم غنی شده با سولفورفان باعث افزایش طول عمر سوسک از 15 به 30٪ (در شرایط خاص) می شود.
00: 20: 34 - اهمیت التهاب کم برای طول عمر.
00: 22: 05 - سبزیجات کریستالی و پودر تخم گشنیز بروکلی به نظر می رسد که انواع مختلف نشانگرهای التهابی را در انسان کاهش دهند.
00: 23: 40 - بازپخش ویدئویی: سرطان، بخش های پیری
00: 24: 14 - مطالعات ماوس نشان می دهد سولفورفان ممکن است در بهبودی سیستم ایمنی سازگاری را بهبود بخشد.
00: 25: 18 - سولفورفان باعث بهبود رشد مو در مدل موشك پستان شد. تصویر در 00: 26: 10.
00: 26: 30 - آغاز بخش مغز و رفتار.
00: 27: 18 - تأثیر عصاره پرتقال بروکلی بر اوتیسم.
00: 27: 48 - اثر گلوکورافامین بر اسکیزوفرنیا.
00: 28: 17 - شروع بحث افسردگی (مکانیزم قابل اعتماد و مطالعات).
00: 31: 21 - مطالعه ماوس با استفاده از مدل های مختلف 10 از افسردگی ناشی از استرس نشان می دهد سولفورفان به طور مشابه به عنوان فلوکستین (پروزاک) موثر است.
00: 32: 00 - مطالعه نشان می دهد که مصرف مستقیم گلوکورافامین در موش ها در جلوگیری از افسردگی از مدل استرس شکست اجتماعی موثر است.
00: 33: 01 - آغاز بخش نابودسازی.
00: 33: 30 - سولفورفان و بیماری آلزایمر.
00: 33: 44 - بیماری سولفورفان و پارکینسون.
00: 33: 51 - بیماری سولفورفان و Hungtington.
00: 34: 13 - سولفورفان باعث افزایش پروتئین شوک حرارت می شود.
00: 34: 43 - شروع بخش آسیب مغزی آسیب دیده.
00: 35: 01 - سولفورفان بلافاصله پس از تزریق TBI تزریق می شود (مطالعه ماوس).
00: 35: 55 - سولفورفان و پلاستیک نرون.
00: 36: 32 - سولفورفان باعث بهبود یادگیری در مدل دیابت نوع II در موش می شود.
00: 37: 19 - دیستروفی عضلانی سولفورفان و دوئن.
00: 37: 44 - مهار مایواستاتین در سلول های ماهواره ای عضلانی (in vitro).
00: 38: 06 - ضبط ویدئویی بعدی: مرگ و میر و سرطان، آسیب DNA، استرس اکسیداتیو و التهاب، دفع بنزن، بیماری قلبی عروقی، دیابت نوع II، اثرات مغز (افسردگی، اوتیسم، اسکیزوفرنی، تولید نئوپروز)، مسیر NRF2.
00: 40: 27 - افکار در مورد بدست آوردن دوز از جوانه های بروکلی یا سولفورفان.
00: 41: 01 - داستان های تخیلی در منزل.
00: 43: 14 - در دمای آشپزی و فعالیت سولفورفان.
00: 43: 45 - تبدیل باکتری های قارچی سولفورفان از گلوکورافین.
00: 44: 24 - مکمل ها بهتر است در ترکیب با میروسیناز فعال سبزیجات کار کنند.
00: 44: 56 - تکنیک های آشپزی و سبزیجات cruciferous.
گرمایش فعالیت پروتئین Epithiospecifier را کاهش می دهد و سازنده سولفورفان را در بروکلی افزایش می دهد
چکیده
سولفورافان، ایزوتیوسیانات کلم بروکلی، یکی از قویترین مواد ضد سرطان مشتق شده از مواد غذایی است. این ترکیب در سبزی دست نخورده وجود ندارد، بلکه از پیش ساز گلوکوزینولات آن، گلوکورافانین، در اثر عمل میروزیناز، آنزیم تیوگلوکوزیداز، در هنگام خرد شدن یا جویدن بافت بروکلی به وجود می آید. با این حال، تعدادی از مطالعات نشان دادهاند که بازده سولفورافان از گلوکورافانین کم است و یک آنالوگ نیتریل غیر فعال زیستی، سولفورافان نیتریل، محصول هیدرولیز اولیه است که بافت گیاه در دمای اتاق خرد میشود. شواهد اخیر نشان می دهد که در آرابیدوپسیس، تشکیل نیتریل از گلوکوزینولات ها توسط یک پروتئین حساس به حرارت، پروتئین epithiospecifier (ESP)، یک کوفاکتور غیر کاتالیزوری میروزیناز کنترل می شود. هدف ما بررسی اثرات حرارت دادن گلچه ها و جوانه های کلم بروکلی بر تشکیل سولفورافان و سولفورافان نیتریل بود، برای تعیین اینکه آیا کلم بروکلی حاوی فعالیت ESP است یا خیر، سپس تغییرات وابسته به گرما در فعالیت ESP، محتوای سولفورافان و زیست فعالی را که با القای اندازه گیری می شود، مرتبط کنیم. فاز دوم آنزیم سم زدایی کوئینون ردوکتاز (QR) در کشت سلولی. گرم کردن گلچه های کلم بروکلی تازه یا جوانه های کلم بروکلی تا دمای 60 درجه سانتیگراد قبل از همگن سازی، به طور همزمان باعث افزایش تشکیل سولفورافان و کاهش تشکیل نیتریل سولفورافان می شود. کاهش قابل توجهی از فعالیت ESP به موازات کاهش تشکیل نیتریل سولفورافان بود. حرارت دادن به دمای 70 درجه سانتیگراد و بالاتر باعث کاهش تشکیل هر دو محصول در گلچه های کلم بروکلی شد، اما نه در جوانه های بروکلی. القای QR در سلولهای هپاتوم lclc7 کشتشده موش به موازات افزایش تشکیل سولفورافان است.
گلها و جوانه های کلم بروکلی قبل از گرم کردن تا دمای 60 درجه سانتیگراد به طور قابل توجهی تشکیل سولفورافان (SF) کاتالیز شده توسط میروسیناز را در عصاره های بافت گیاهی پس از خرد کردن افزایش داد. این امر با کاهش تشکیل سولفورافان نیتریل (SF نیتریل) و فعالیت پروتئین سازنده اپی تسیوس (ESP) همراه بود.
در نتیجه، سولفورافان یک ماده شیمیایی گیاهی است که در کلم بروکلی و سایر سبزیجات چلیپایی یافت می شود. مقدار کنترل نشده اکسیدان ناشی از عوامل داخلی و خارجی می تواند باعث استرس اکسیداتیو در بدن انسان شود که در نهایت ممکن است منجر به انواع مشکلات سلامتی شود. سولفورافان می تواند تولید Nrf2 را فعال کند، یک فاکتور رونویسی که به تنظیم مکانیسم های آنتی اکسیدانی محافظ که پاسخ سلول به اکسیدان ها را کنترل می کند، کمک می کند. دامنه اطلاعات ما محدود به مسائل مربوط به کایروپراکتیک و سلامت ستون فقرات است. برای بحث در مورد موضوع، لطفاً از دکتر خیمنز بپرسید یا با ما تماس بگیرید915-850-0900.
درد پشتاین یکی از مهمترین دلایل معلولیت و روزهای از دست رفته در کار در سراسر جهان است. کمردرد دومین دلیل شایع مراجعه به مطب توسط پزشکان است که بیشتر از عفونت های دستگاه تنفسی فوقانی است. تقریباً 80 درصد مردم حداقل یک بار در طول زندگی خود درد کمر را تجربه خواهند کرد. ستون فقرات یک ساختار پیچیده است که از استخوان ها ، مفاصل ، رباط ها و ماهیچه ها در میان سایر بافت های نرم تشکیل شده است. به همین دلیل ، صدمات و یا شرایط وخیم مانند دیسک های فتق دیسک، در نهایت می تواند منجر به علائم درد پشت شود. آسیب های ورزشی یا آسیب های ناشی از تصادفات خودرو اغلب اغلب علت درد پشت هستند، اما گاهی اوقات ساده ترین حرکات می توانند نتایج دردناکی داشته باشند. خوشبختانه، گزینه های درمان جایگزین، مانند مراقبت از کیهان پراکسی، می تواند به کاهش درد در استفاده از تنظیمات ستون فقرات و دستکاری دست کمک کند و در نهایت بهبود تسکین درد را کاهش دهد.
IFM's Find A Practitioner بزرگترین شبکه ارجاع در پزشکی کاربردی است که برای کمک به بیماران در یافتن پزشکان طب عملکردی در هر نقطه از جهان ایجاد شده است. با توجه به تحصیلات گسترده در پزشکی کاربردی ، پزشکان مجاز IFM اولین بار در نتایج جستجو ذکر شده اند